Chapitre 1 :
Structure des acides nucléiques..
I Structure primaire.
Un acide nucléique est composé d’une base, d’un sucre et d’un phosphate.
A Les bases.
On a deux familles de bases :
· Les bases à noyaux puriques.
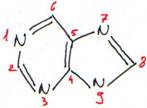
Ce type de noyau est à la base de l’adénine (A) ou 6-aminopurine (NH2 en 6) et de la guanine (G) ou 2-amino,6oxydépurine (NH2 en 2 et =O en 6).
· Les bases à noyaux pyrimidiques.
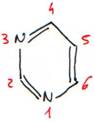
Ce type de noyau est à la base de l’uracile (U) ou 2-6dioxyde pyrimidine (=O en 2 et 6), de la thymidine (T) ou 2-6dioxyde 5méthylpyrimidine (=O en 2 et 6 et CH3 en 5) et la cytosine (C) ou 2oxyde 4aminopyrimidine (=O en 2 et NH3 en 4).
On assiste à une désamination oxydative de la cytosine en uracile. C’est une réaction fréquente.
Dans l’ADN, l’uracile sera réparé par des enzymes de réparation car la présence de cette base entraînerait trop de mutation.
Au contraire, dans l’ARN, ces changements ne sont pas graves car la durée de vie de l’ARN et des protéines est courte.
L’absence de T permet la reconnaissance de l’ARN pour les enzymes de dégradation et un gain d’énergie (la greffe de la fonction méthyle demande de l’énergie).
B Les sucres (le ribose et le désoxyribose).
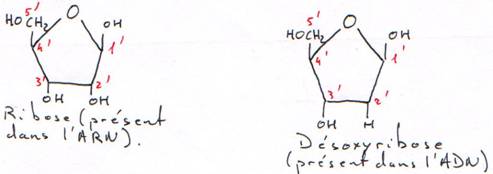
Le désoxyribose est une forme beaucoup plus stable que le ribose. Cette forme désoxy entraîne la stabilisation de l’hélice.
C Le phosphate (fonction acide).
Les phosphates permettent la solubilisation de l’ADN dans l’eau grâce à leurs charges négatives.
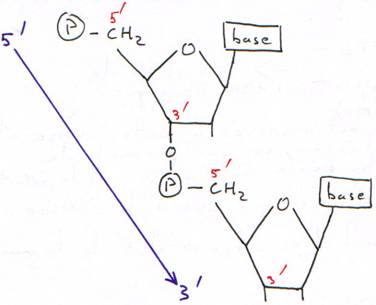
Base + sucre = nucléoside
Nucléoside + phosphate = nucléotide.
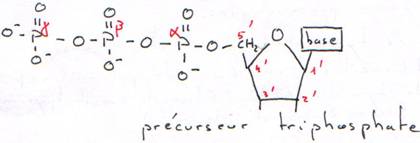
Les phosphates permettent la polymérisation des acides nucléiques (des nucléotides).
D Les modifications des bases.
Sur l’ADN et sur l’ARN, on trouve des bases modifiées après la synthèse ou après la réplication. Les modifications de l’ARN sont toujours post-transcriptionnelles.
Exemple de modifications par méthylation.
· On trouve beaucoup de cytosine modifiée (méthylée) chez les eucaryotes. Ces modifications n’affectent pourtant pas leur appariement.
Quand l’ADN est très méthylé, il ne sera pas transcrit. Dans certaines cellules, par méthylation, on peut contrôler la transcription de l’ADN : c’est une régulation en fonction du type de cellules.
· Pendant la réplication :
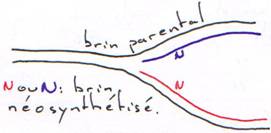
Après la synthèse de l’ADN, on assiste à une correction sur épreuve du brin néosynthétisé. La reconnaissance parentale est possible car cet ADN est plus méthylé que le jeune brin.
· Les bactéries ont un moyen de se protéger des virus : elles synthétisent des enzymes qui digèrent l’ADN (DNAses de restriction) en coupant sur une séquence particulière.
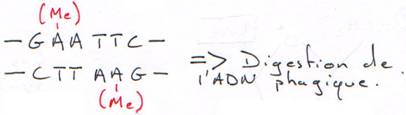
Une méthylase est associée à cette DNAse et va méthyler l’ADN bactérien, ce qui la protège de ses propres enzymes. Bien sûr, la méthylase va agir avant la DNAse.
II Structure secondaire.
A L’ADN.
L’ADN est formé de deux brins antiparallèles dont les bases sont hydrophobes.

Les couples de bases (A-T et C-G) sont maintenus grâce à des interactions hydrogènes. Ces interactions permettent l’association et la structure en hélice.
Entre les noyaux aromatiques, on a des interactions par liaisons p.
|
Type
|
Nombre de bases par tour
|
Sens de rotation
|
Angle entre chaque base
|
|
B
|
10
|
Dextrogyre
|
36°
|
|
A
|
11
|
Dextrogyre
|
|
|
C
|
9,3
|
Dextrogyre
|
|
|
Z
|
12
|
Lévogyre
|
|
La forme Z peut exister expérimentalement, mais elle est moins probable dans la nature. Toutefois, la synthèse d’anticorps anti-Z met en évidence la présence de cette forme spéciale.
On trouve souvent l’ARN avec une structure secondaire.

III Le surenroulement.
L’ADN non-linéaire peut, en général, être considéré comme circulaire.
Le surenroulement entraîne un compactage de l’ADN. Pour retirer cet enroulement, il faut libérer un brin (couper un des deux brins). On passe alors à une forme circulaire ouverte (forme relaxée). Si l’on coupe le second brin, on obtient une forme linéaire.
Les enzymes gyrases (ou topo-isomérases II) coupent un brin et l’amène à effectuer un super-tour négatif (consommation d’ATP).
La revergyrase fait effectuer à l’ADN un super-tour positif.
La topo-isomérase I coupe un brin, attend qu’il se déroule puis ressoude la coupure. Cette enzyme fonctionne pendant la réplication de l’ADN.
Le rôle du surenroulement :
- L’ADN est beaucoup plus compact.
- Le surenroulement négatif aide à la séparation des brins alors que le surenroulement positif resserre les brins.
- Il régule l’expression génique en permettant ou non la transcription de la partie surenroulée.
IV Dosage et purification.
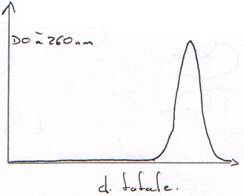
Les bases absorbent les UV vers 260 nm. (A : 259 nm ; G : 253 nm ; C : 271 nm).
e = 10 000 M-1cm-1 (DO = e.[C].l = 104.[C])
Le poids moyen d’un nucléotide monophosphate est de 300g/L.
On a alors : DO = (104/300).[C] (C est en g/L).
Pour des oligonucléotides, de 10 à 20 bases, cette formule fonctionne.
Pour une solution d’ADN, il faut 50 µg/mL pour avoir 1 de DO.
Pour une solution d’ARN, il faut 40 µg/mL pour avoir 1 de DO.
L’énergie est en parti prise pour maintenir la structure, donc, l’absorption diminue.
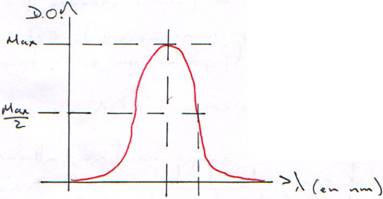
Quand la DO (à l=260 nm) est égale à 2×DO (à l=280 nm), c’est que la solution ne contient que de l’ADN.
L’utilisation des colorants.
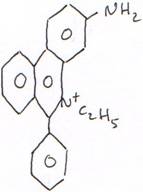
Exemple avec le bromure d’éthidium (B Et) : c’est un composé fluorescent, excité à 254 nm, il émet vers le rouge. Il se place entre les bases et fonctionne mieux sur l’ADN que sur l’ARN. C’est un agent très mutagène.
V Purification.
A Fractionnement cellulaire.
Pour les cellules eucaryotes, l’ADN est dans le noyau, l’ARN est dans le cytoplasme et l’ADN mitochondrial est dans les mitochondries.
On a trois composants principaux : le noyau, le cytoplasme et les mitochondries.
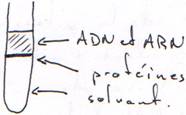
B La séparation des acides nucléiques des protéines.
In vivo, l’ADN est entouré de protéines (les histones et des enzymes), comme l’ARN. Pour les séparer, on dénature les protéines (avec du SDS ou du guanadium) et on effectue la séparation grâce à un solvant organique (phénol, chloroforme).
C La concentration par précipitation.
On prépare une solution d’acides nucléiques, d’éthanol et d’un sel monovalent (Na+Cl-).
Le sel va rentrer en compétition avec les molécules d’eau et l’éthanol.
Les acides nucléiques vont alors précipiter et on obtiendra une solution relativement pure.
D Séparation des acides nucléiques.
On fait une digestion enzymatique de l’ADN ou de l’ARN par un DNase ou par une RNase.
E Les centrifugations.
1 La centrifugation différentielle.
Cette centrifugation donne une séparation grossière.
2 La centrifugation isopycnique (à l’équilibre).
On utilise une solution saline qui forme un gradient de densité (CsCl). Si on tourne avec d(=r)=1,7, les molécules vont descendre ou monter pour se mettre au niveau de leur densité.

L’ARN est plus dense que l’ADN qui est lui-même plus dense que les protéines.
3 La centrifugation zonale (en fonction de la vitesse de sédimentation).
Les éléments les plus lourds descendent plus vite que les plus légers. On obtient alors la vitesse de sédimentation qui peut servir pour séparer les différents acides nucléiques et les différentes protéines.
On réalise ces centrifugations avec une solution de saccharose et l’unité de mesure est le suedberg.
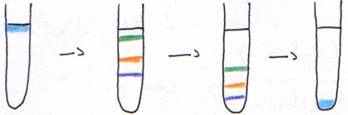
4 Les cas particuliers.
Dans la centrifugation isopycnique, pour voir les acides nucléiques, on ajoute un colorant. Il y a donc modification de la densité : ADN à ADN + B Et et ARN à ARN + B Et. On a alors une diminution de la densité.
Dans un tube, avec du matériel génétique bactérien, on trouve de l’ADN chromosomique, de l’ADN plasmidial et de l’ARN. Dans ce tube, on rajoute aussi le colorant.
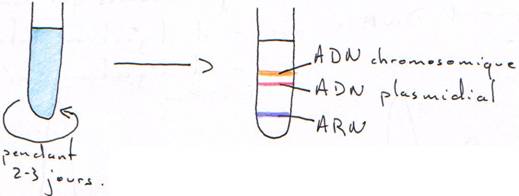
L’ADN plasmidial prend moins de colorant car il est sur enroulé.
F L’électrophorèse.
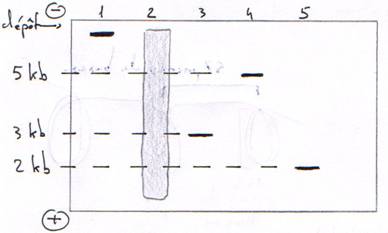
1 : génome procaryote ou eucaryote normal. 2 : ADN fragmenté par une enzyme de restriction. 3 : plasmide bactérien non digéré, sur enroulé donc plus petit. 4 : plasmide coupé une fois. 5 : plasmide coupé deux fois.
L’électrophorèse se fait sur gel d’agarose ou chimique (acrylamide).
Plus les mailles sont grosses, plus les molécules seront ralenties.
La charge donne le sens de migration.
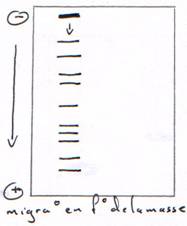
1 : migration des ARN ribosomiques. 2 : avec ARNt. 3 : ARNm. 4 : ARN totaux (on ne voit pas l’ARNm car il n’est pas en assez grand nombre).
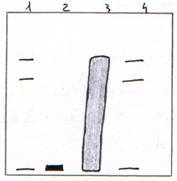
Préparation de chromosomes qui vont être digérés par une DNase en digestion ménagée et en digestion totale.
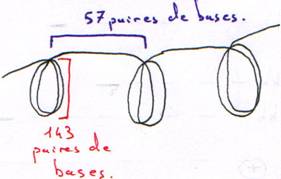
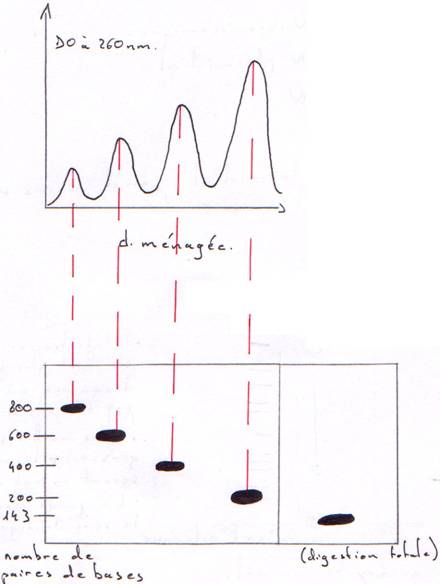
On trouve une répétition tous les 200 nucléotides (143 + 57) : c’est un nucléosome.
Chapitre 2 - Partie 1 :
Dénaturation et hybridation..
I Dénaturation.
On va retirer la structure secondaire ou primaire de l’ADN ou de l’ARN. On chauffe pour que les deux brins se séparent (à la température de fusion ou Tm : melting). Le nombre de liaisons H-H et la composition du milieu influencent ce Tm :
- Le nombre de liaisons H-H dépend de la longueur du fragment (jusqu’à 150 liaisons). Ce nombre est important pour les petits fragments (50 à 60 bases).
- La composition en bases GC / AT.
- La présence de mis-appariement.
- la composition du milieu : une augmentation de sels monovalents élève le Tm. Une augmentation d’urée, de formamide permet de diminuer le Tm.
- Un pH extrême diminue le Tm.
Quand on dénature de l’ADN, on change ses propriétés.
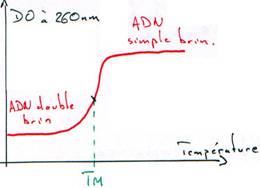
Sa densité augmente ; en centrifugation isopycnique, l’ADN simple brin est au niveau de l’ARN.
- Les colonnes d’hydroxyapatite (phosphate de calcium) retiennent l’ADN double brin et laissent passer le simple brin.
- Les membranes de nylon ou de nitrocellulose retiennent le simple brin et laissent passer le double brin.
L’hybridation dépend de la température, de la concentration et du temps.
II Hybridation.
A Hybridation en phase liquide.
L’ADN génomique qui va être cloné est marqué radioactivement.
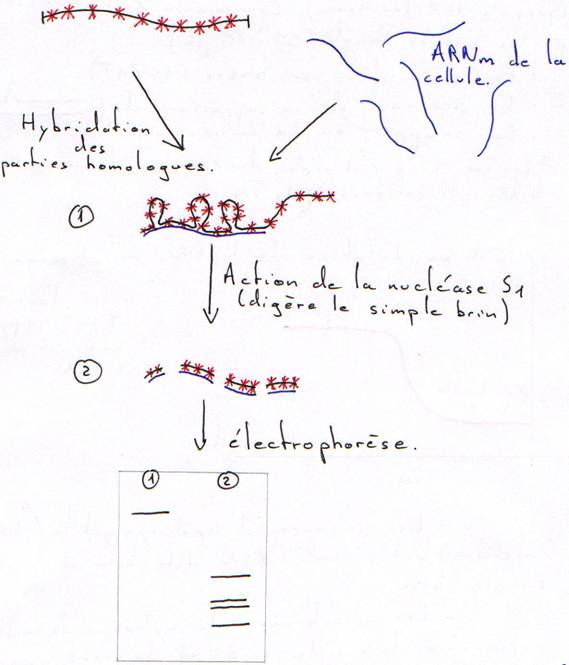
Cette électrophorèse permet de voir la taille des brins hybridés.
Remarque : Pour une même concentration d’ADN, plus le génome est petit (ou peu complexe), plus la renaturation est rapide.
Dans un tube, on met 10µg/mL d’ADN, on chauffe puis on laisse refroidir.
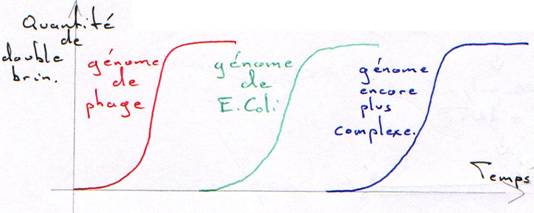
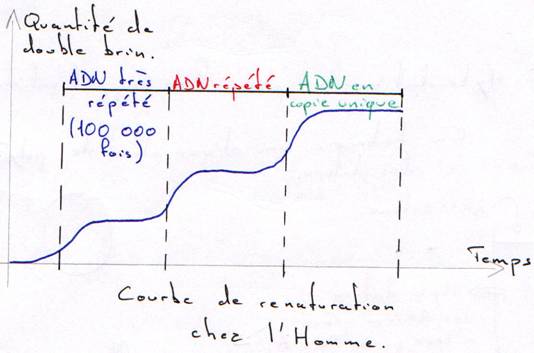
On a une courbe en paliers : ce sont les trois populations différentes d’ADN dans le même génome.
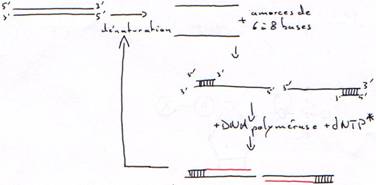
B Hybridation d’une amorce.
L’amorce est un oligonucléotides synthétisé chimiquement.
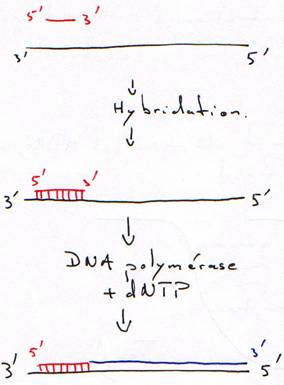
C Hybridation sur phase fixe (support solide).
On fabrique un oligonucléotide polyT que l’on fixe sur des billes.

D Fixation de l’ADN sur une membrane de nitrocellulose ou de nylon.
Remarque : ces deux types de membranes retiennent les ADN simple brin et les ARN.
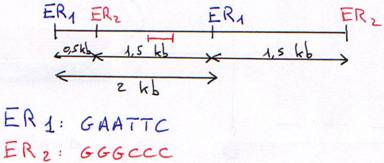
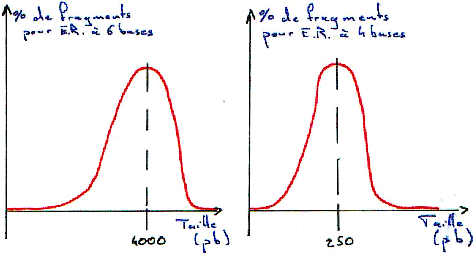
1 Technique de Southern.
On fait digérer l'ADN (normal) par des enzymes de restriction (coupure en moyenne toutes les 4000 bases). On travaille toujours à Tm.


L'ADN simple brin va se fixer sur le support soluble.
On prend un morceau d'ADN (d'1 kb) marqué radioactivement (une sonde) puis on va regarder où la sonde s'hybride.

La sonde va alors s'hybrider sur le support solide. On lave ensuite la solution puis on fait une révélation par auto-radiographie.
Si la sonde est un oligonucléotide, on a deux cas:
- l'électrophorèse présente une seule bande pour chaque enzyme de restriction: on est en présence d'un seul gène.
- l'électrophorèse présente deux bandes pour chaque enzyme de restriction: on alors deux gènes.

2 Technique de Northern.
On travail à température proche de Tm avec hybridation sur milieu solide : gel sur lequel on dépose des ARNm.
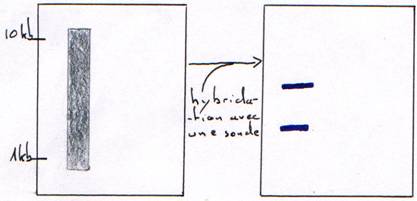
Le résultat nous permet de connaître la taille de l'ARN.
Chapitre 2 - Partie 2 :
Marquage des acides nucléiques.
Le marquage le plus simple est le marquage radioactif. On peut réaliser celui-ci avec différents isotopes.
- L’isotope le plus classique est le 32P, d’une demi-vie de 14 jours et E=1,7 MeV (très forte sensibilité).
- Le 35S : sa demi-vie est de 90 jours et E=0,17 Mev. Il est moins sensible mais plus précis que le 32P.
- Le 3H, sa demi-vie est de 12 ans et E=0,02 Mev.
Exemple avec hybridation sur chromosome.

Pour placer un 35S, on l’insère à la place d’un O.
Pour les marquages froids, on rajoutera des morceaux que l’on peut colorer. Pour les marquages radioactifs, on peut partir de nucléotides triphosphates marqués.
· Translation de coupure ou Nick translation.
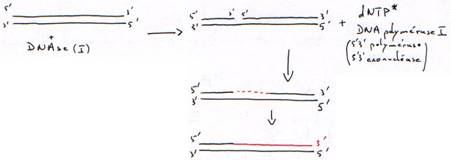
- Le phosphate doit être marqué en a car les phosphates b et g seront sortis pendant la synthèse.
- Les deux brins seront marqués.
· Technique de Random Priming (amorçage au hasard).
On part d’ADN double brin en forte concentration.
· Marquage d’ADN double brin grâce à la RNA polymérase.

Dans le cas de deux promoteurs différents sur le même génome, on a les résultats suivants :
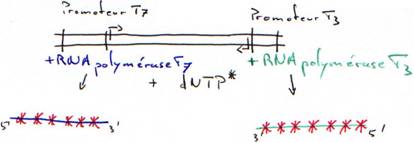
Pour la transcription, le promoteur donne le sens de transcription et le brin transcrit.
On peut marquer un acide nucléique en lui ajoutant un phosphore marqué radioactivement sur une extrémité.
- On fait d’abord agir une phosphatase pour enlever le phosphore non marqué.
- On fait en suite agir une kinase avec de l’ATP marqué en g. Le marquage se fera en 5’.
Chapitre 2 - Partie 3 :
Les outils du génie génétique.
I Les enzymes.
On va se servir des enzymes pour couper, coller et synthétiser des acides nucléiques.
A Les polymérases.
Toutes les polymérases agissent de 5’ vers 3’. En cas d’erreur, il faut réparer de 5’ vers 3’ pour permettre la correction et la re-synthèse.
1 Les DNA polymérases.
a Les DNA polymérases DNA dépendantes.
Ces DNA polymérases fabriquent de l’ADN à partir d’une matrice d’ADN. On trouve trois activités chez ces enzymes :
- Synthèse de 5’ vers 3’ à partir d’une amorce hybridée (matrice simple brin hybridée)
- Activité 3’5’ exonucléase.
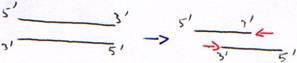
L’activité polymérase est dix fois plus rapide que l’activité exonucléase. Cette dernière sert donc à diminuer la quantité d’erreurs : c’est une activité de correction de la DNA polymérase.
- Activité 5’3’ exonucléase.

Cette activité est une activité de réparation.

Cette activité permet de réparer plus que ce qui est nécessaire : elle permet ainsi le renouvellement de l’ADN.
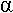 La DNA polymérase I de E. coli.
La DNA polymérase I de E. coli.
Elle possède les trois activités précédentes. C’est une enzyme de 100 kDa, composée de deux fragments (un de 70 kDa et un autre de 30 kDa).
Le fragment qui possède les activités 5’3’ polymérase et 3’5’ exonucléase est appelé fragment de Klenow.
 Les DNA polymérases T4 et T7.
Les DNA polymérases T4 et T7.
Ces enzymes ne possèdent que les activités 5’3’ polymérases et 3’5’ exonucléase. Quand la T7 est modifiée, elle n’a plus que l’activité polymérase.
 La Taq DNA polymérase.
La Taq DNA polymérase.
La Taq est la DNA polymérase de Thermus aquaticus (bactérie vivant à 80°C). Cette enzyme ne possède que la fonction polymérase. Toutefois elle peut synthétiser en continue et à haute température sans être dénaturée.
Application de la Taq :
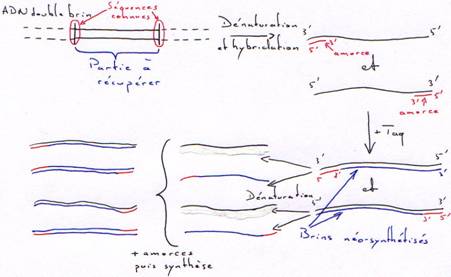
On obtient rapidement une amplification de la partie du brin comprise entre les amorces. Au bout du 40ème cycle et à partir d’un seul exemplaire, on obtient 1012 copies.
A partir d’un génome complet, on peut extraire une molécule bornée des deux côtés par les amorces et en obtenir un très grand nombre.
b Les DNA polymérases ARN dépendantes (les reverse transcriptases).
On trouve ce type de polymérases chez les rétrovirus et dans les cellules eucaryotes mais en faible quantité.
Ces DNA polymérases ont une activité 5’3’ polymérase et nécessitent une amorce pour synthétiser ; elles ont souvent une activité de coupure de l’ARN quand il est hybridé (RNase H).
On purifie un ARNm (polyadénylé) par une hybridation avec un oligo-dT.
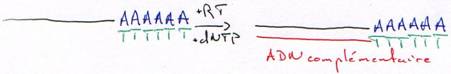
c DNA polymérase terminal-transférase.
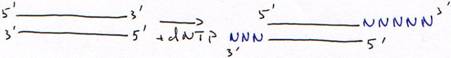
Ajout d’un seul nucléotide :
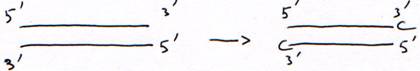
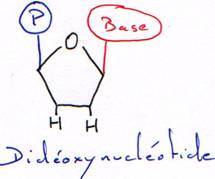
Avec un acide aminé sans fonction OH en 3’, la synthèse est arrêtée au bout d’un nucléotide. Cet acide aminé sans fonction OH est dit didéoxynucléotide.
B Les enzymes de dégradation.
1 Les RNA polymérases.
Les RNA polymérases agissent au niveau de la transcription. Elles synthétisent de 5’ vers 3’. Elles reconnaissent l’ADN double brin ainsi qu’un promoteur qu’il porte (site de reconnaissance et site de départ).

La transcription peut être dans les deux sens. Ce sens est définie par le promoteur.
2 Les DNAses.
Les DNAses sont des enzymes qui digèrent l’ADN. Il existe aussi des RNAses et des nucléases.
aDNase à reconnaissance non spécifique.
La DNase la plus connue est la DNase I qui reconnaît l’ADN double brin et qui le coupe (chez E. coli).
La nucléase S1 ne coupe que l’ADN simple brin.

b Les DNAses de restriction.
Ce sont des enzymes qui digèrent l’ADN double brin sur des séquences spécifiques. Elles reconnaissent des palindromes.

Fragment reconnu par l’enzyme de restriction Eco R1 de E. coli.
Ces enzymes de restriction donnent deux types de coupures : des coupures franches (au même niveau sur les deux brins) et des coupures à extrémités sortantes.
3 Les RNAses.
Les RNAses vont couper l’ARN.
· La RNase A : elle coupe le simple brin en 3’ des résidus pyrimidiques (après C et U) et donne des nucléotides 3’phosphate.
· La RNase T1 : elle coupe du simple brin après G et donne un 3’phosphate.
· La RNase H : chez les rétrovirus, elle élimine les brins d’ARN rétro-transcrits. Elle coupe après tous les nucléotides
4 Ligase.
· La T4 DNA ligase fonctionne sur de l’ADN double brin avec au moins, une liaison OH-Phosphore.
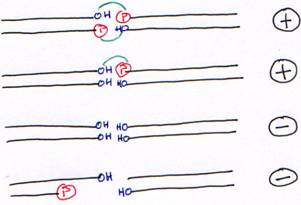
· La T4 RNA ligase lie un ARN (du simple brin) avec son extrémité 5’ phosphatée sur une extrémité 3’ hydroxylée. On pourra mettre de l’ADN avec l’extrémité 5’ phosphatée.
5 Phosphatase, kinase et méthylase
· Les phosphatases catalysent le retrait du groupement phosphate en 5’.
· Les kinases avec la T4 polynucléotide-kinase : elle rajoute un phosphate en 5’.
· Les méthylases vont particulièrement agir sur les sites de restriction en les méthylant pour empêcher l’action des enzymes de restriction.
II Les vecteurs.
Les vecteurs sont capables de transporter un fragment d’ADN et de l’amplifier par auto réplication.
A Les plasmides.
1 La transformation.
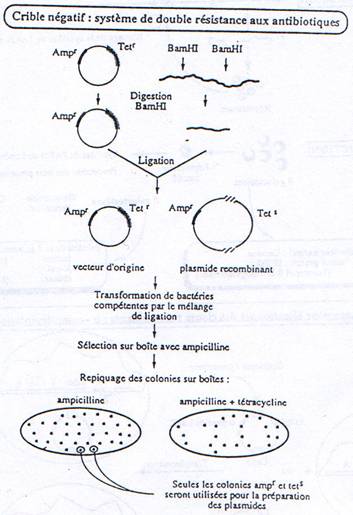
Il faut connaître les parties indispensables pour utiliser le plasmide comme vecteur.
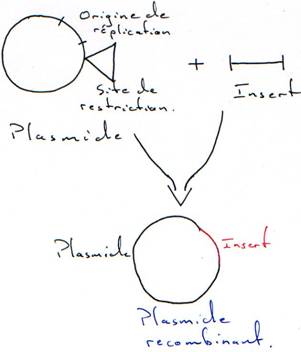
On a besoin d’un gène de résistance à un antibiotique (en général à l’ampicilline) pour pouvoir effectuer une sélection.
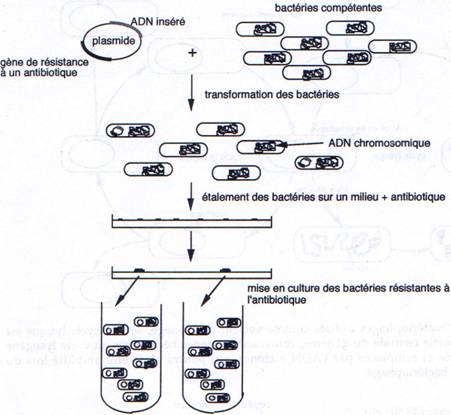
A la place d’avoir un seul site de restriction, on peut en avoir un second.
-GAATCC GAATCC- : c’est un polylinker.
On place, sur le plasmide, un gène qui code pour une enzyme qui colorera la bactérie.
L’intérêt d’un tel crible (coloré) est que l’on peut vérifier que le plasmide ne s’est pas religué seul.
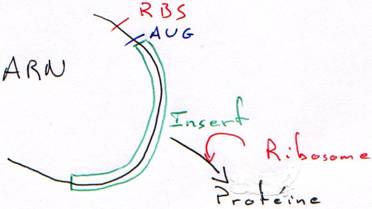
Quand on place un insert, le plasmide est recombinant. La RNA polymérase va pouvoir transcrire cet insert. Toutefois, ce dernier n’est en phase qu’une fois sur trois. Quand il n’est pas en phase, il n’y a pas de synthèse de protéine.
2 Les promoteurs des RNA polymérases.
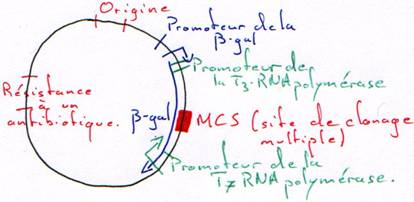
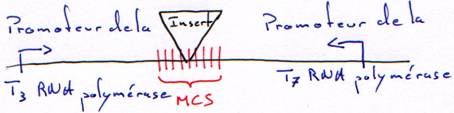
Le vecteur est un morceau d’ADN qui permet d’amplifier la synthèse de l’ADN à transcrire.
B Les phages.
Les phages sont des virus à bactéries dont on peut isoler la descendance.
1 Le phage 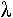 (lambda).
(lambda).
Le phage l est un virus à ADN double brin, linéaire de 50 kb. Au milieu du génome, on trouve une partie de 20 kb qui n’est pas indispensable au cycle lytique. Elle peut être remplacée par un ADN étranger.

Le phage l est encapsidé, il reconnaît un récepteur au maltose, s’y fixe et injecte son ADN qui sera utilisé pour la transformation.
On est capable de fabriquer ce virus in vitro : protéines + ADN + encapsidation à virus
On peut aussi sélectionner un phage recombinant de phages non-recombinants.
· Méthode de préparation du phage l :
On retire d’abord le fragment de 20 kb inutile et on obtient un bras droit de 20 kb et un bras gauche de 10 kb.
On sépare les deux fragments par centrifugation zonale.
On ajoute l’ADN recombinant que l’on va fixer par ligation.
Remarque : dans la capside, il ne rentre qu’un fragment d’ADN que 50 kb (+ ou – 5 kb).
Quand il rentre dans la bactérie, il devient circulaire avec extrémités auto-complémentaires. Sa réplication est comme celle d’un plasmide.
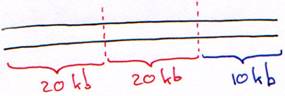
2 Le phage M13.
C’est un phage de 6 kb à ADN simple brin. Il rentre dans les cellules par les pili sexuels. Quand l’ADN est rentré, il passe en double brin puis commence à se répliquer comme un plasmide mais la réplication devient continue sur un seul brin : réplication en rolling circle. On obtient des ADN simples brins qui seront empaquetés et qui sortiront de la cellule.
Ce phage accepte de plus gros fragment d’ADN : on peut rajouter des sites de clonage multiple (MCS)
C Les systèmes hybrides.
1 Plasmide + origine de M13.
Ici, M13 est helper : il sert à produire les enzymes nécessaires à la synthèse de l’ADN simple brin.
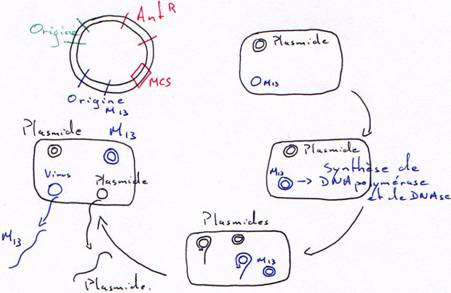
2 Les cosmides : plasmide + extrémités cos (cohésives) de (lambda).
Les cosmides sont des vecteurs hybrides portant à la fois :
- Des séquences d’origine phagique permettant leur encapsidation in vitro (séquence cos).
- Des séquences d’origine plasmidique : gène de résistance à un antibiotique et séquence permettant au plasmide de se répliquer dans une bactérie comme un simple plasmide (séquence ori).
Ceci permet de se dispenser des régions essentielles de l’ADN du phage et de construire des ADN recombinants portant jusqu’à 45 kb d’ADN étranger.
3 Les YAC (yeast artificial chromosome) ou chromosome de levure.
Ces vecteurs de clonages peuvent se présenter sous deux formes : une forme circulaire, permettant sa manipulation chez E. Coli (et en particulier l’insertion d’ADN exogène et son amplification è ils possèdent une séquence ori et un gène de résistance à un antibiotique) et une forme linéaire, obtenue après coupure par BamHI et EcoRI et qui permet d’obtenir les deux “bras” du YAC. Ces deux bras contiennent les télomères (TEL), une origine de réplication levure (ARS) un centromère (CEN) ainsi que des gènes (URA et TRP) utilisés pour la sélection des clones. La ligation de ces bras à un très grand fragment d’ADN humain (100 à 1000 kb) extrait de cellules en culture le transforme en pseudo chromosome de levure capable après introduction dans ce microorganisme de se répliquer (grâce à ARS), de stabiliser ses extrémités (grâce aux séquences TEL) et de se répartir entre les cellules filles lors de la division cellulaire (grâce à la séquence CEN).

III Les oligonucléotides.
On peut synthétiser des oligonucléotides jusqu’à 50 mères (ou bases). La synthèse se fait de 3’ vers 5’.
On greffe un nucléotide sur un support puis on rajoute un nucléotide protégé contre la fixation non-désirée d’un autre nucléotide à la fois. Entre chaque ajout d’un nucléotide, on enlève la protection. Finalement, on décroche l’oligonucléotide et obtient la séquence voulue ou variable.
A Le clonage.
Le génome humain comporte 3.109 paires de bases.
Pour en étudier une partie, on fait un tri que l’on va amplifier.
On fabrique des banques d’ADN génomique et des banques d’ARN. On transforme ces ARN en une population de clones. Une banque de cDNA est une banque génomique représentant l’ensemble du génome de la cellule de départ qui est découpé en fragments de taille connue.
Pour l’étude des ARNm, on dispose de deux techniques.
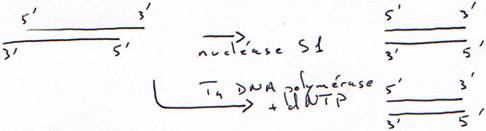
1 Technique à la nucléase S1.
On transforme l’ARN en ADN. On va donc hybrider cet ARN avec un oligo-dT.
Finalement, on obtiendra une copie qui n’est pas totale car elle aura perdu une partie son extrémité 5’ afin d’avoir des extrémités franches.
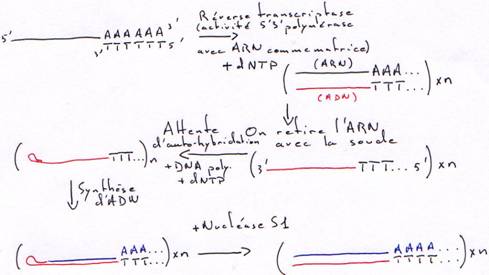
2 Technique à la RNase H.
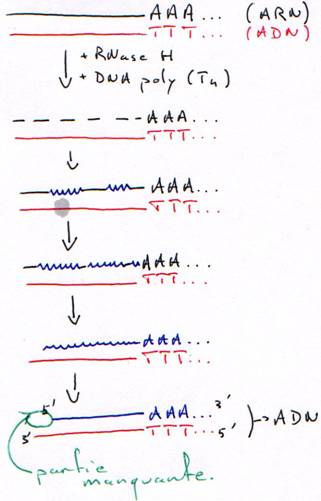
La DNA polymérase ne peut pas synthétiser le début car il n’y a pas d’amorce.
Dans ce cas aussi, il manquera un bout.
è Les cDNA ne sont pas complets : la séquence est aléatoire en fonction du lieu de la coupure.
B Utilisation des cDNA.
1 cDNA avec vecteur (et ligase).
La coupure est réalisée avec une enzyme de restriction (souvent avec Sau 3A) en digestion ménagée. On récupère alors des fragments dont la taille est proche de 20 kb.
Par centrifugation zonale (6 heures à 30 000 tours/minutes), les molécules sont séparées selon leur taille.
On réalise ensuite une électrophorèse pour vérifier la taille des fragments. On sélectionne ensuite les sections selon leur taille. On en trouve qui se recoupent car on utilise plusieurs génomes pour réaliser la digestion.
Les fragments sont ensuite insérés dans un vecteur qui est souvent le phage l. On garde son bras gauche de 10 kb et son bras droit de 20 kb. On rajoute l’insert de 20 kb, de la T4 DNA ligase et de l’ATP.
Le phage l ainsi modifié va être encapsidé si la taille du nouveau total est proche de 50 kb. Une fois que le phage a infecté les bactéries, on va réaliser un criblage de ces banques.
a Criblage avec antibiotiques.
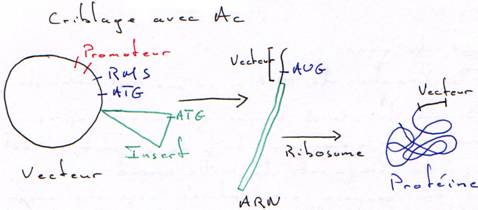
b Criblage Immunologique.
Tous les clones de la banque seront traduits. On sélectionnera les mutants qui fixeront l’anticorps correspondant à la protéine de l’insert.
2 Ajout d’un linker.
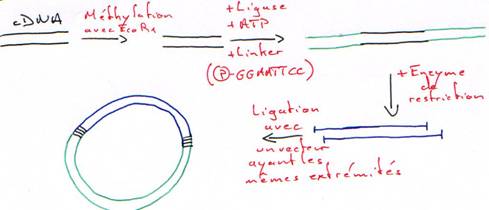
Chapitre 2 - Partie 4 :
Séquençage des acides nucléiques.
Connaître la séquence primaire des acides nucléiques est une étape déterminante pour la mise en évidence de fonctions éventuelles codées par un ARN ou un fragment d’ADN cloné reconnaissance de séquences consensus, prédiction de séquences peptidiques, détermination de structures secondaires,
I Séquençage de l’ARN.
On utilise des endonucléases spécifiques ayant une spécificité pour un certain type de base:
RNase T1 : clive spécifiquement l’extrémité 3’ des résidus G
RNase U2: clive spécifiquement l’extrémité 3’ des résidus A
RNase PhyM : dive spécifiquement l’extrémité 3’ des résidus A et U
RNase 13C : clive spécifiquement l’extrémité 3’ des résidus C et U.
Ces ribonucléases sont utilisées sur un ARN marqué. à des concentrations telles que l’on ait une seule coupure par molécule et ce, statistiquement à chacune des positions possibles. On libère ainsi des fragments de taille variables, clivés spécifiquement, reflétant la position des bases. L’analyse s’effectue sur un gel d'acrylamide dénaturant permettant de séparer des fragments différents de un nucléotide en taille.
N.B. : Un gel séquence se lit du + vers le - = de 5’ vers 3’ si on marque l’ARN en 5’ ou sur toute la longueur et de 3’ vers 5’ si on marque en 3’.
II Séquençage de l’ADN.
A Méthode de Maxam et Gilbert.
1 Préparation de l’ADN
Marquage des extrémités 5’ du fragment à cloner.
Séparation des deux extrémités marquées.
2 Clivage chimique de l’ADN.
Le principe de la méthode consiste à modifier spécifiquement par un réactif chimique un type de base et de cliver la chaîne d’ADN au niveau de ce nucléotide modifié. Les agents modifiant sont utilisés en concentration telle que l’on aura qu’une seule coupure par molécule, et ce à toutes les positions possibles.
Les réactifs utilisés sont
-> DMS (diméthylsulfate) - méthylation des purines puis 0,1 M NaOH -> coupe après G puis 0,1 M HC1 -> coupe après A -> hydrazine - coupe après C et T -> hydrazine + NaCl -> coupe après C.
L’analyse et la lecture du gel sont identiques à celles décrites pour le séquençage de l’ARN.
B Méthode de Sanger.
Pour cette technique, il est nécessaire de disposer d’une amorce (court enchaînement de déoxynucléotides) ayant une séquence de bases complémentaires à une partie de la molécule d’ADN à étudier. Comme cette séquence n’est en général pas connue, on choisit cette amorce dans le vecteur de clonage (dont la séquence est connue).
1 Préparation de l’ADN.
L’ADN à étudier doit être sous la forme simple brin pour permettre l’hybridation de cette amorce. Pour cela, on a deux possibilités :
- dénaturation (les résultats sont moins bons car une partie des plasmides à séquencer va se ré hybrider)
- si le vecteur de clonage possède une origine de réplication d’un bactériophage simple brin (fd ou m13 par exemple), il suffit de surinfecter par ce bactériophage pour obtenir une grande quantité de notre plasmide sous la forme simple brin.
2 Etape d’hybridation/élongation.
L’extension de la chaîne continue jusqu’à l’incorporation d’un didéoxynucléotide (pas de OH en 2’ ni en 3’ du ribose) à la place du déoxynucléotide correspondant, ce qui provoque la terminaison de la chaîne. Le rapport déoxynucléotide/didéoxynucléotide est choisi de façon à obtenir statistiquement un arrêt à chacune des positions du nucléotide étudié. Quatre essais sont donc réalisés (rajout soit du ddATP, soit du ddCTP ou du ddGTP ou du ddTTP). Chaque tube contient donc en fin de réaction une population de fragments de taille variable, ayant tous la même extrémité 5’ et se terminant tous en 3’ par le même didéoxynucléotide à toutes les positions possibles.
L’analyse et la lecture du gel sont identiques à celles décrites pour le séquençage de l’ARN (le marquage se fait soit en 5’ de l’amorce, soit en réalisant l’élongation avec un dNTPa32P).
Chapitre 2 - Partie 5 :
Mutagenèse dirigée.
Pour aller plus loin dans la compréhension de la structure et de la fonction des gènes, on doit disposer de moyens permettant de modifier la séquence de l’ADN et d’analyser l’effet de ces changements sur la fonction des gènes. Pour modifier la séquence de l’ADN, on utilise la technique de mutagenèse in vitro de gènes clonés.
Il y a deux grands types de mutagenèse : la mutagenèse aléatoire et la mutagenèse dirigée. Nous ne présenterons ici que deux exemples de mutagenèse dirigée.
I Mutagenèse par extension enzymatique d’une amorce synthétique portant la mutation.
Dans cette méthode, on fabrique un oligonucléotide (par synthèse chimique) qui contient la mutation flanquée de 10 à 15 nucléotides de séquence non mutée. Cet oligonucléotide “mutagène” est hybridé à la séquence complémentaire sauvage présente dans l’ADN monocaténaire préparé à partir d’un clone dans un phage ou un phagemide. Il se forme donc un ADN hétéroduplexe. Cet oligonucléotide sert alors d’amorce pour la synthèse enzymatique d’ADN in vitro par une polymérase qui convertit l’ADN monocaténaire sauvage en une forme bicaténaire. L’ADN hétéroduplexe ainsi obtenu contient donc un brin sauvage et un brin muté. Cet ADN est ligaturé et transformé chez E. coli. Lors de la première réplication, on obtient donc un plasmide sauvage et un plasmide muté (qui se répliqueront à leur tour pour donner un mélange de plasmides sauvages et mutants). Pour discriminer ces deux types de plasmides (ou enrichir les cultures en clones mutants), un grand nombre de méthodes sont possibles : nous étudierons ici une de ces techniques : la mutagenèse dirigée selon Kunckel.
L’ADN matrice simple brin est préparé dans une souche d’E. coli déficiente en uracile déglycosidase (ung-), de manière à pouvoir contenir plusieurs résidus uracile à la place de thymine. L’oligonucléotide mutagène est apparié et est utilisé comme amorce pour la synthèse “in vitro” d’un brin complémentaire de la matrice. Cette synthèse est faite en présence des quatre désoxyribonucléotides normaux. Après ligature, les molécules hétéroduplexes sont introduites dans une souche ung+ d’E. CoU. Une fois dans la cellule, le brin de type sauvage (la matrice) est attaqué par l’uracile déglycosidase, qui provoque des coupures du brin d’ADN et ce brin est dégradé avant de pouvoir être répliqué. Comme le brin qui contient la mutation ne contient pas d’uracile, il n’est pas attaqué et il est répliqué normalement. On obtient ainsi essentiellement des plasmides mutants. Un séquençage permet de vérifier la présence de la mutation.
II Mutagenèse par PCR.
Deux segments d’ADN, l’un s’étendant “en amont” et l’autre “en aval” de la mutation désirée sont amplifiés en utilisant des oligonucléotides qui introduisent la mutation désirée et génèrent des produits de PCR qui se chevauchent sur 20 nucléotides ou plus dans la région de la mutation (étape 1 et 2). Quand ces deux produits de PCR sont mélangés et hybridés l’un avec l’autre, un des produits possibles est composé de deux brins d’ADN qui s’hybrident sur une courte région au niveau de leur extrémité 3’(étape 3). Cette molécule peut être polymérisée par l’ADN polymérase pour générer une matrice correspondant au fragment entier portant la mutation (étape 4). Cette matrice est ensuite amplifiée par une troisième PCR utilisant les oligonucléotides 1 et 4.
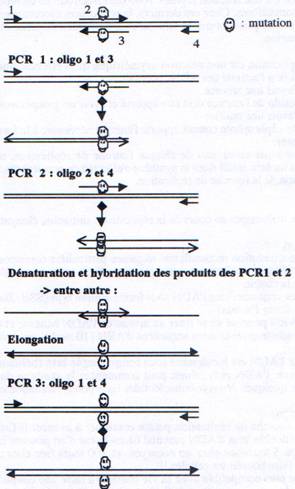
Chapitre 3 - Partie 1 :
Réplication.
I Généralités.
A Les expériences de Taylor (1957- incorporation de thymidine 3H) et de Meselson et Stahl (1958-incorporation de 15N) ont permis de démontrer que la réplication s’effectue par un mécanisme semi-conservatif (les deux autres hypothèses impliquaient des modèles conservatif ou dispersif).
B En 1960, par une expérience de Pulse-Chase en thymidine 3H, Cairns a montré que la réplication s’effectuait de manière bidirectionnelle à partir d’une (procaryotes) ou de plusieurs origines de réplication (eucaryotes : le nombre d’origines de réplications dépend de la taille du génome à répliquer - la phase S dure environ 2h - et peut atteindre plusieurs milliers). Chaque origine de réplication est donc constituée de deux fourches de réplication.
La réplication est une réaction rapide : 1000 nucl./sec/fourche de réplication chez les bactéries et 50 nucl./sec chez les mammifères. Chez ces derniers, la réplication s’accompagne de la synthèse protéique des histones qui s’assemblent pour former la structure chromatinienne. Cette étape supplémentaire ralentit la vitesse de polymérisation.
Enfin, la réplication est une réaction asymétrique qui s’effectue uniquement dans le sens 5’à3’. Cette asymétrie est liée à l’activité des ADNs polymérases :
- nécessité d’avoir une amorce ;
- le dernier résidu de l’amorce doit être apparié et avoir un groupement hydroxyle libre (3’OH) ;
- nécessité d’avoir une matrice ;
- le nucléotide triphosphate entrant apporte l’énergie nécessaire à la formation de la liaison phosphodiester.
On distingue ainsi au niveau de chaque fourche de réplication, un brin précoce qui subit une synthèse continue et un brin tardif dont la synthèse s’effectue de manière discontinue dans un sens opposé à celui de la progression de la fourche de réplication.
II Mécanisme.
On distingue trois étapes au cours de la réplication : initiation, élongation, terminaison.
A L’initiation.
Une protéine d’initiation reconnaît une séquence particulière correspondant à l’origine de réplication.
Une ADN hélicase se fixe sur cette protéine d’initiation et va dérouler la double hélice en se déplaçant le long de la chaîne.
Des protéines vont stabiliser 1’ADN sous forme simple brin (SSB : Single Strand Binding protéine ou HDP : Helix Destabilizing Protéine)
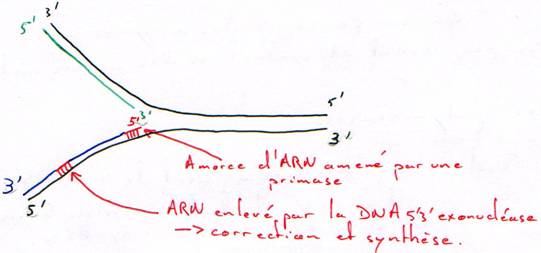
Enfin, une ARN primase va se fixer au niveau de l’ADN hélicase et constituer ainsi le primosome. Cette enzyme va synthétiser de courtes séquences d’ARN (10 nucl.) qui vont par la suite servir d’amorce à l’ADN polymérase.
Une fois que l’ADN est localement sous forme simple brin (hélicase et SSB), et que la primase a synthétisé une amorce, l’ADN polymérase peut commencer la synthèse du brin complémentaire au brin parental. Au bout de quelques déoxyribonucléotides incorporés, l’initiation est terminée.
B L’élongation.
Pour que la fourche de réplication puisse continuer à avancer il faut dérouler la double hélice. Il faudrait que chaque double brin d’ADN parental (dans le cas d’un génome linéaire) tourne sur lui même à une vitesse d’environ 5 tours/sec chez un eucaryote et 100 tours /sec chez un procaryote. L’énergie ainsi libérée reviendrait à faire bouillir les cellules !!!
Une solution plus compatible avec la vie consiste à faire une coupure sur l’un des deux brins de la matrice, à laisser ce brin tourner autour de l’autre puis à le raccrocher ; c’est le rôle des topo isomérases de type I.
Dans le cas des génomes circulaires, c’est l’intervention de deux topo isomérases de type II (qui coupe les deux brins d’ADN) dont la gyrase qui va permettre de séparer les deux brins parentaux.
Ces problèmes de contraintes structurales réglés, l’ADN polymérase va pouvoir synthétiser en continu à partir d’une chaîne parentale un nouveau brin d’ADN, le brin précoce.
Par contre sur l’autre chaîne parentale, la synthèse s’effectue de manière discontinue. La primase va régulièrement synthétiser des amorces d’ARN à partir desquelles l’ADN polymérase va synthétiser les fragments d’Okasaki. Ces fragments sont reliés les uns aux autres par l’ADN ligase, après que l’ADN polymérase ait dégradé l’amorce ARN (grâce à son activité exonucléase 5’à3’) puis re-synthétisé la partie manquante.
C La terminaison.
Dans le cas d’un ADN circulaire (procaryotes), la terminaison est réalisée lorsque les deux fourches de réplication se rencontrent (la topo isomérase de type IV assure l’étape de ligation).
Pour l’ADN linéaire, les fourches de réplication s’arrêtent soit lorsqu’elles rencontrent une autre fourche de réplication, soit lorsqu’elles arrivent à l’extrémité d’un chromosome.
Pour les génomes linéaires eucaryotes, une télomérase permet d’ajouter quelques nucléotides à l’extrémité du brin tardif, de manière à ne pas raccourcir les chromosomes à chaque réplication.
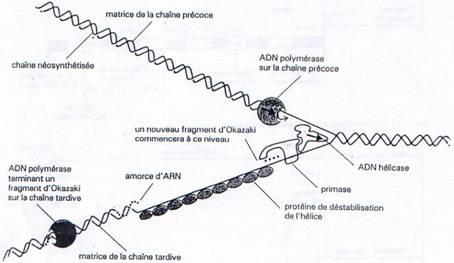
Chapitre 3 - Partie 2 :
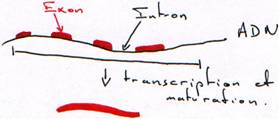
Transcription.
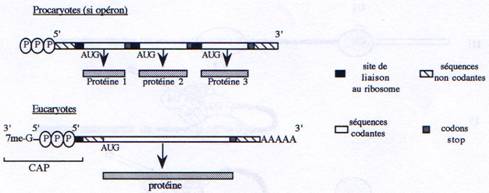
Chez les procaryotes, un gène est défini structurellement comme une séquence d’ADN comprenant un promoteur, un site d’initiation et un site de terminaison. Chez les eucaryotes, il faut compléter cette définition par la présence d’introns localisés à l’intérieur de la partie transcrite du gène. D’autre part, il existe chez les procaryotes des gènes organisés en opérons (voies métaboliques) et donnant des ARNm polycistroniques. Mais on trouve également des gènes de structure plus simple ne contenant, comme chez les eucaryotes, qu’une seule unité de traduction.
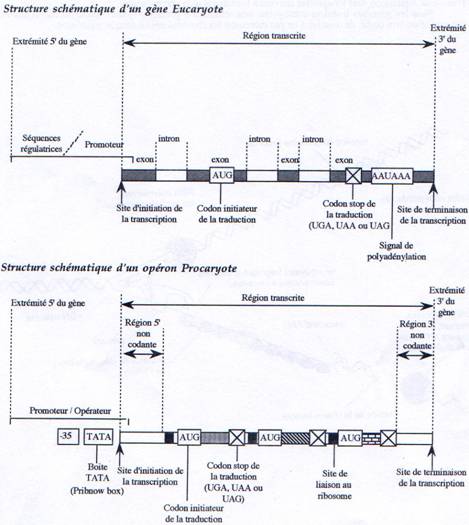
L’unité de transcription d’un gène correspond à la séquence présente dans le transcrit primaire d’ARN.
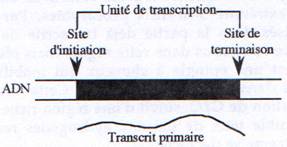
Chez les procaryotes comme chez les eucaryotes, la transcription est divisée en trois étapes initiation, élongation et terminaison.
I Transcription chez les procaryotes.
L’ARN polymérase ADN dépendante est une enzyme formée de plusieurs sous-unités le core-enzyme a la structure a2bb’ et l’holoenzyme a2bb’s chez E. coli. Elle polymérise les nucléotides dans le sens 5’à3’ à partir d’un promoteur. Cette enzyme n’a pas besoin d’amorce, n’utilise qu’un brin comme matrice et progresse à une vitesse d’environ 30 nucl./sec.
A L’initiation.
L’initiation de la transcription se fait au niveau d’une séquence appelée promoteur. Ce promoteur est constitué de deux séquences très conservées, situées respectivement 35 et 10 pb en amont du site d’initiation de la transcription (+1).

Le core-enzyme a une affinité faible pour les séquences d’ADN double brin. Par contre la présence du facteur s dans l’holoenzyme confère à l’ARN polymérase une très forte affinité pour le promoteur. L’holoenzyme explore donc l’ADN par liaison non spécifique et se lie fortement aux régions promotrices qu’elle dénature localement pour commencer la transcription. L’initiation va durer le temps que la polymérase associe 7 ou 8 ribonucléotides sous forme d’un polymère hybridé au brin matrice. Comme le core-enzyme présente une très forte affinité pour les hétéroduplexes ARN/ADN, le facteur s se décroche et c’est le core-enzyme qui va seul continuer la transcription.
B L’élongation.
Le core-enzyme continue la polymérisation tout en déroulant l’ADN en aval et en le ré enroulant une fois la séquence copiée. En amont du site de polymérisation on trouve un hybride ARN/ADN d’environ 17pb.
C La terminaison.
Aucune homologie de séquence n’a été retrouvée sur l’ADN au-delà du dernier nucléotide présent à l’extrémité 3’ d’ARN procaryotes. Par conséquent, les signaux de terminaison sont localisés dans la partie déjà transcrite de l’ARN. D’autre part, on ne retrouve pas de séquence consensus dans cette région mais plutôt une structure conservée.
Cette structure est une épingle à cheveux qui mobilise les nucléotides de l’ARN normalement impliqués dans l’hybride ARN/ADN. Cette structure secondaire contient en général une forte proportion de G/C suivit d’une région riche en U. Le raccourcissement de l’hétéroduplexe et le faible taux de liaisons hydrogènes restant déstabilisent l’hybride ARN/ADN et le core-enzyme se décroche.
Certains terminateurs possèdent trop peu de G/C dans la région correspondant à l’épingle à cheveux pour que cette structure ne se forme pas. Un facteur supplémentaire (facteur r) va alors stabiliser cette structure secondaire. On parle dans ce cas de terminaison r dépendante par opposition aux terminaisons r indépendantes.
D Les ARNs polycistroniques.
Certains gènes procaryotes sont rassemblés en opérons structures qui renferment plusieurs gènes, impliqués dans une même voie métabolique, sous le contrôle d’un même promoteur. On retrouve donc au sein d’un même ARN, des séquences codant pour plusieurs protéines.
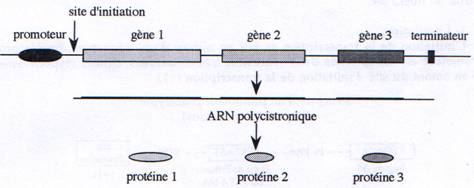
II Transcription chez les eucaryotes.
Il existe chez les eucaryotes trois ARN polymérases différentes:
ARN polymérase 1 : transcription des ARN ribosomiques
ARN polymérase 2 : transcription des ARN messagers et ARNsn
ARN polymérase 3 : transcription des ARN de transfert et des ARNr 5S.
Ces ARN polymérases sont de grosses protéines multimériques présentant des propriétés différentes.
Chez les eucaryotes le mécanisme de base de la transcription est identique à ce qui a été décrit pour les procaryotes. Cependant, la structure des promoteurs est différente et les transcrits primaires obtenus sont toujours monocistroniques. Enfin, une des différences majeures concerne les modifications post-transcriptionnelles des ARN eucaryotes;
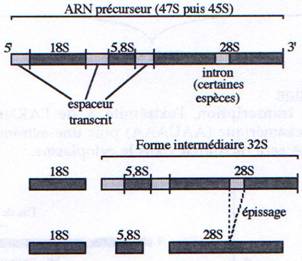
ARNr (sauf le 5S) : méthylation, bases modifiées (par ex : UàyU-pseudoU) et épissage
ARNt : méthylation, épissage et bases modifiées (UàWU)
ARNr 5S : pas de modification
ARNm : coiffés, épissés et polyadénylés. Certaines adénines peuvent également être méthylées
A Maturation des ARNr.
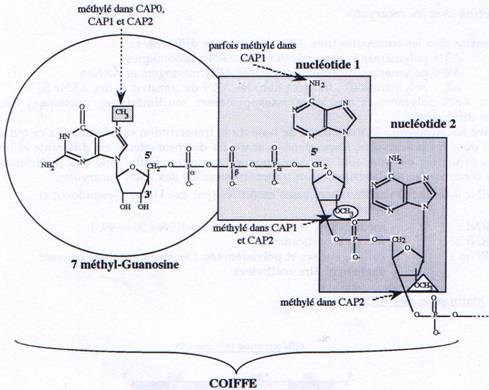
B Capping.
Au cours de la transcription, lorsque les ARNs atteignent 20 à 30 nucléotides de longueur, une 7 méthyl-guanosine est ajoutée sur le premier nucléotide en 5’, par une liaison phosphodiester 5’à5’. Tous les ARNm sont coiffés excepté les ARNm codant pour les histones. Il existe trois types de coiffes que l’on distingue en fonction de leur taux de méthylation
- CAPO : seule la guanosine est méthylée en position 7
- CAP1 : CAPO + méthylation du C2’ du premier nucléotide et parfois de l’hétérocycle
- CAP2 : CAP1 + méthylation du CT du second nucléotide
C Polyadénylation.
A la fin de la transcription, l’extrémité 3’ de l’ARNm est clivé au niveau d’une séquence conservée hexamérique (AAUAAA) puis une extrémité poly A est rajoutée. Par la suite cette queue polyA sera raccourcie dans le cytoplasme.
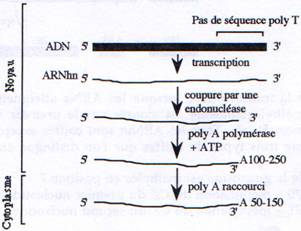
D Epissage.
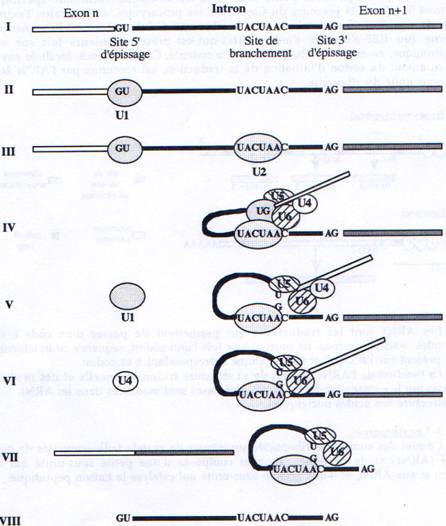
A la fin de la transcription, alors que le polyA vient d’être rajouté, les introns vont être éliminés. Les introns font donc partie du transcrit primaire et sont absents de l’ARN mature. Le mécanisme d’épissage se fait grâce à l’intervention de particules ribonucléo-protéiques appelées les snRNP (U1 à U6). Chacune de ces particules est formée d’un ARN et de plusieurs protéines. Un type d’épissage est décrit dans la figure ci dessous :
I : Structure du transcrit primaire
II : U1 reconnaît le site 5’ d’épissage par interaction ARN/ARN ;
III : U2 reconnaît le site de branchement par le même type d’interaction ;
IV : l’hétérotrimère U4/U5/U6 se lie. U5 reconnaît le site 5’ d’épissage. U6 interagit
avec U2.
V : U1 se dissocie. U5 se déplace de l’exon à l’intron.
VI : U4 se dissocie. U6 catalyse la trans-estérification - le site 5’ d’épissage est coupé et le lasso est formé.
VII : Le site 3’ d’épissage est coupé et les deux exons ligaturés. L’ARN épissé est libéré.
U2/U5/U6 restent accroché sur le lasso.
VIII : Le lasso est “débranché”
Chapitre 3 - Partie 2 :
Traduction.
I Les différents partenaires de la traduction.
A L’ARNm.
L’ARNm est la matrice à partir de laquelle s’effectue la traduction des séquences nucléotidiques en séquences peptidiques chez les eucaryotes comme chez les procaryotes. Chez les procaryotes, transcription et traduction sont deux mécanismes couplés puisque ayant lieu dans un seul compartiment cellulaire. A l’inverse chez les eucaryotes, la traduction a lieu uniquement dans le cytoplasme. D’autre part, les modifications post-transcriptionnelles (capping et polyadénylation) des ARNm eucaryotes sont importantes pour la localisation du transcrit et l’efficacité de la traduction.
Chez les eucaryotes, la petite sous-unité du ribosome reconnaît spécifiquement l’extrémité 5’ grâce à la présence du Cap. Chez les procaryotes, au contraire, l’extrémité 5’ n’a pas de signification particulière pour le ribosome. Celui-ci reconnaît la séquence Shine-Dalgarno (ou RBS-Ribosome Binding Site) qui est présent plusieurs fois sur un ARN polycistronique, en amont de chaque séquence codante. Cette séquence, localisée environ 8 à 10 nucléotides en amont du codon d’initiation de la traduction, est reconnue par l’ARNr 16S de la petite sous-unité du ribosome.
B Les ARNt.
Les ARNt sont les traducteurs qui permettent de passer d’un code à l’autre : nucléotides à acides aminés. Ils portent à la fois l’anti-codon, séquence complémentaire du codon présent sur l’ARNm, et l’acide aminé correspondant à ce codon.
La fonction de l’ARNt dépend de sa structure tridimensionnelle et des modifications des bases qui le constituent. Environ 10% des bases sont modifiées dans les ARNt. (Voir structure des acides nucléiques)
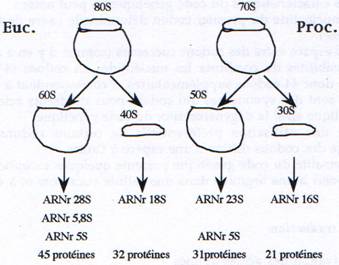
C Les ribosomes.
Ce sont des complexes ribonucléo-protéiques de grande taille composés de molécules d’ARN (ARNr) et de protéines. Ils sont composés d’une petite sous-unité qui se lie à l’ARNm et aux ARNt, et d’une grande sous-unité qui catalyse la liaison peptidique.
II Principe de la traduction.
Tout comme la réplication et la transcription, la synthèse protéique est polarisée. Les ribosomes se déplacent dans le sens 5’ vers 3’ sur l’ARNm, et synthétisent le polypeptide correspondant de l’extrémité NH2 terminale vers l’extrémité COOH terminale.
La séquence de l’ARNm est décodée par groupe de trois nucléotides (codon) qui correspondent à un acide aminé particulier ou aux signaux d’initiation et de terminaison. L’ensemble de cette codification constitue le code génétique dont la correspondance nucléotides ó acides aminés est représentée dans le tableau suivant :
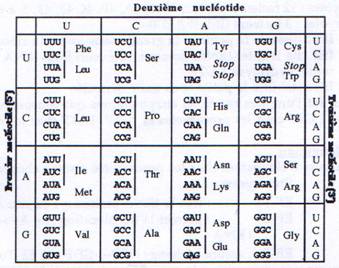
Parmi les principales caractéristiques du code génétique on peut noter:
- Le premier nucléotide du premier codon détermine le cadre de lecture ouvert (ORF : Open Reading Frame)
- Il n’y a pas d’espace entre des codons successifs (comme il y en a entre deux mots)
- Il y a 64 possibilités de combiner les nucléotides en codons (43). Comme il y a 20 acides aminés, il y a donc 44 codons supplémentaires. 3 correspondent à des codons stop ou non-sens, les autres sont des synonymes qui codent pour différents acides aminés (théorie du Wobble). On explique ainsi la dégénérescence du code génétique.
- On observe une utilisation préférentielle de certains codons pour une espèce donnée. Ainsi l’usage des codons diffère d’une espèce à l’autre.
- Enfin l’universalité du code génétique présente quelques exceptions. Par exemple, le codon AGA correspond à une arginine dans une cellule eucaryote et à un codon stop dans une mitochondrie.
III Le mécanisme de la traduction.
A L’activation des acides aminés.
Il existe au moins 20 aminoacyl tRNA synthétases qui vont permettre la charge des acides aminés sur les ARNt
Ac. Aminé + ATP à Ac. Aminé-AMP +PPi
Ac. Aminé -AMP + tRNA à Ac. Aminé-tRNA + AMP
B L’initiation.
A la différence de la réplication ou de la transcription dans lesquelles on observe une polymérisation durant l’initiation, il n’y a pas de formation de liaison peptidique au cours de cette étape dans la traduction.
Un seul codon sert d’initiateur dans 99% des traductions : AUG.
On retrouve parfois le codon GUG comme initiateur.
De nombreux facteurs vont contrôler et stabiliser l’initiation
Eucaryotes : 12 facteurs (elF1, 2, 2B, 3, 4A, 4B, 4C 4E, 4F, 5, 6 et p220)
Procaryotes : 3 facteurs (IF1, IF2, IF3)
IF1 : dissocie la petite et la grande sous-unité du ribosome
IF2 : lie le fMet-tRNA, favorise la liaison fMet-tRNA sur la petite sous-unité, hydrolyse 1 GTP
IF3 : stabilise la petite sous-unité dissociée
La traduction est l’une des réactions enzymatiques qui consomme le plus d’énergie. L’initiation nécessite 1 GTP chez les procaryotes et 1 GTP/1 ATP chez les eucaryotes.
C L’élongation.
Il existe 3 facteurs d’élongation chez les procaryotes comme chez les eucaryotes :
|
Eucaryotes
|
Procaryotes
|
|
EF1a
|
EF-Tu : fixe le tRNA-aminoacyl au site A
|
|
EF2
|
EF-G : permet la translocation site A à site P et éjecte le tRNA
|
|
EF1bg
|
EF-Ts : assure l’échange GTP à GDP sur EF-Tu
|
L’énergie consommée pour un acide aminé incorporé est de 1 GTP pour la charge du tRNA-aminoacyl et 1 GTP dans la translocation. La vitesse d’élongation est de 1 à 3 acides aminés par sec. chez les eucaryotes et 15-20/sec. chez les procaryotes.
Très peu d’erreurs sont commises au cours de l’élongation: 1/100 000 erreurs au cours de la translocation, 1/1000 à 1/10 000 erreurs d’incorporation d’un aminoacide. Enfin le ribosome rend l’hybridation codon/anticodon 100 fois plus fidèle qu’en solution et 20 000 fois plus stable...
D Terminaison.
Il n’existe qu’un seul facteur de terminaison chez les eucaryotes (RF : Releasing Factor) et trois chez les procaryotes (RF1, RF2, RF3).
RF1 reconnaît les codons UAG et UAA au site A
RF2 reconnaît les codons UGA et UAA au site A
RF3 éjecte RF1 et RF2 du site A en hydrolysant 1 GTP.
Au total la synthèse d’une seule protéine de 100 acides aminés chez un procaryote, aura nécessité l’hydrolyse de 100 ATP (charge de l’acide aminé sur les ARNt) et 202 GTP !!!!
Chapitre 1 :
I Généralités.
On peut dire que l’on a du vivant quand une cellule peut donner deux cellules identiques de manière autonome.
L’information est l’ADN ; il doit être dupliqué ainsi que l’enveloppe (membrane + phospholipides). Doivent être synthétisées, des protéines pour la synthèse de l’enveloppe (phospholipides) et pour la synthèse de l’ADN (informations).
Le plus petit génome procaryote connu est composé de 2.10^6 pb. 260 gènes devraient être nécessaires pour le métabolisme de base.
Chez E. Coli, on trouve 4,2.10^6 pb, soit, 4800 gènes.
Lorsque la distance entre deux gènes est entre 4 et 5 acides nucléiques, on a une structure compacte
On peut aussi avoir des gènes polycistroniques.
A Les levures (eucaryotes).
Les levures ont un noyau et des organites. Les mitochondries vont fournir de l’ATP grâce à la respiration. Le génome mitochondrial est composé de 2.10^7 pb, soit, 6200 gènes. La levure a des chromosomes. Les procaryotes n’ont qu’une molécule d’ADN alors que les eucaryotes en ont plusieurs (chromosomes).
B La drosophile.
Le génome de la drosophile est constitué de 10^8 pb. Une cellule va donner des millions de cellules mais différenciées.
C Le génome humain.
Le génome humain est composé par 3.10^9 pb, soit, 50 000 gènes.
D Les batraciens.
Chez les batraciens, on trouve 10^11 paires de bases.
E Les parasites.
Parmi les parasites, on trouve : les mycoplasmes(2.10^5 pb) et les phages (ex : le phage de 50 000 pb).
1 Les phages.
Une fois le phage dans la bactérie, selon l’espèce, il a différent devenir :
- L’ADN phagique peut s’insérer dans l’ADN bactérien.
- L’ADN va servir à produire du phage : à un faible niveau, il n’entraîne pas la mort de la bactérie ; à un haut niveau, il entraîne la mort de la bactérie.
2 Les virus.
On trouve des virus à ADN ou à ARN. Les virus à ADN ont une taille variable comprise entre 4000 pb et 10^5 pb. Parmi les virus à ARN (rétrovirus), on trouve le HIV (14.10^3 nt) et le virus de la grippe.
3 Les plasmides.
Les plasmides sont de petites molécules d’ADN autonome qui vivent dans les bactéries. En général, on utilise des plasmides de l’ordre de 4000 pb. Il existe toutefois de plus gros plasmides qui peuvent aller jusqu’à 10^5 pb. Ils possèdent tous une séquence « Ori » qui leur permet une réplication autonome.
On a deux familles de plasmides en fonction « d’Ori » :
- de 3 à 5 copies par bactérie,
- plus de 100 copies par bactérie.
Les résistances aux antibiotiques sont dues aux plasmides (gènes de résistance).
F Temps de réplication et chromosome.
Chez E. Coli, le temps de génération est de 20 minutes (doublement de population). Pour une cellule eucaryote, une mitose a une durée comprise entre 20 et 24 heures.
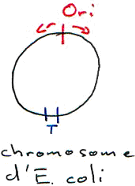
La réplication du chromosome bactérien est bidirectionnelle avec une seule origine de réplication alors que chez les eucaryotes, il y a plusieurs « Ori » le long des chromosomes.
Chez les eucaryotes, centromère et télomères sont des séquences particulières. Les télomères sont reliés au vieillissement : ils forment un système de protection.
Chapitre 2 :
Les outils.
Parmi les outils du génie génétique, on trouve des vecteurs (plasmides, phages, virus) et des enzymes.
I Les enzymes.
A Les exonucléases.
On trouve trois différents types d’exonucléase :
- type I : elles coupent de 3’  5’
5’
- type II : elles coupent de 5’ 3’
- type III : elles coupent de 3’ 5’ et de 5’ 3’.
B Les endonucléases.
1 Principe de fonctionnement.
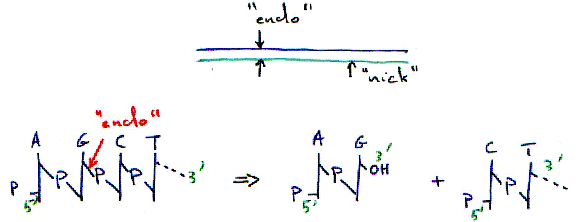
Ici, l’endo-enzyme est non spécifique : elle coupe n’importe où.
2 Les enzymes de restriction.
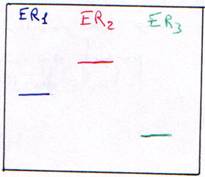
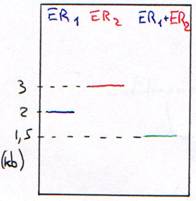
Une enzyme qui coupe à un endroit précis (toujours le même), sur un site de restriction est une enzyme spécifique : c’est une enzyme de restriction (ER). On trouve entre 300 et 350 ER.
Les sites spécifiques sont en général constitués de 4pb (pour 200 ER), ou de 5pb (4 ou 5 ER) ou de 6pb (une trentaine d’ER) ou bien, de 7 à 12pb (quelques unes). Les enzymes de restriction utilisées sont celles qui reconnaissent un site (séquence) et coupe sur ce même site.
3 Digestion de l’ADN.
Si la digestion est complète, on obtiendra les résultant présentés dans les courbes suivantes. Dans ce cas, on se trouve en excès d’enzymes.
Si la digestion n’est que partielle (ou ménagée), les fragments seront plus longs.
C La RNA polymérase II.
L’ARN pré-messager va donner un ARNm dans le cytosol. La famille des ARNu présente des cas petits, impliqués dans le processus de maturation des messagers.
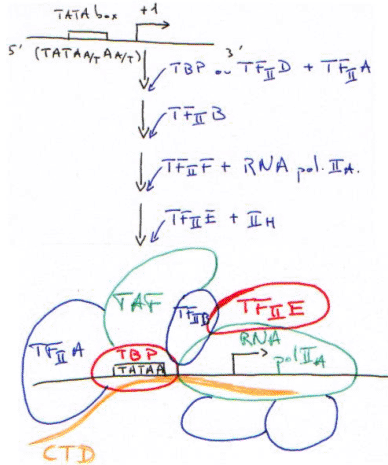
TBP + TAF = TFIID.
CTD : domaine carboxy-terminal de la grande sous-unité (220-240Da) de la RNA polymérase II.
Le TBP reconnaît la TATA-box puis recrute tous les autres facteurs.
La RNA polymérase II (14 sous-unités) interagit avec la TBP par l’intermédiaire du CTD.
Le complexe d’initiation est maintenant fermé.
Le CTD est représenté par 52 fois un heptapeptide (Y S P S T P S : tyrosine, sérine, proline, sérine, thréonine, proline, sérine). C’est un ensemble d’acides aminés phosphorylables par les enzymes sérine-thréonine-kinase et thyrosine-kinase.
Le CTD va se faire phosphoryler par un complexe enzymatique contenu dans TFIIA.
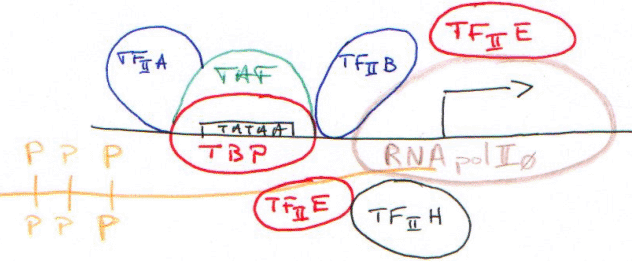
Le complexe d’initiation (CI) est maintenant ouvert : c’est la « clearance » du promoteur.
Quand le domaine carboxy-terminal est phosphorylé, on parle de RNApol II 0 : le CTD phosphorylé n’interagit plus avec la TBP.
TFIIH : ce facteur contient l’élément d’une maladie génétique : xéroderma pigmentosum, qui donne des difficultés à réparer les lésions sur l’ADN.
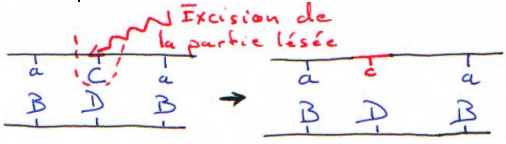
Dans ce TFIIH, il y a une activité hélicase 5’3’ pour la réparation et une activité hélicase 3’5’ pour la transcription.
Dans cette maladie, l’activité de réparation est absente (maladie monogénique).
Ce facteur est composé de 9 sous-unités : c’est un facteur de transcription qui se comporte comme un facteur de réparation.
La kinase du CTD :
TFIIE contrôle l’activité de TFIIH.
In vivo, en réalité, c’est un immense complexe préformé qui reconnaît le promoteur par l’intermédiaire du TBP qui reconnaît la TATA-box. Ce complexe est appelé une « holoenzyme ».
A ce complexe sont aussi associé les facteurs de maturation de l’ARNm. Ces facteurs sont reconnus par des TAF.
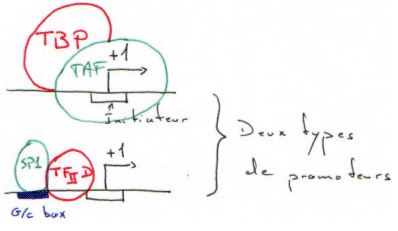
La G/C box est reconnue par le facteur SP1 ; il y a alors ajout de TFIID, puis, on reprend le cycle comme au début.
Les gènes possédant une G/C box sont appelés « gènes de ménage » ou « house keeping genes ».
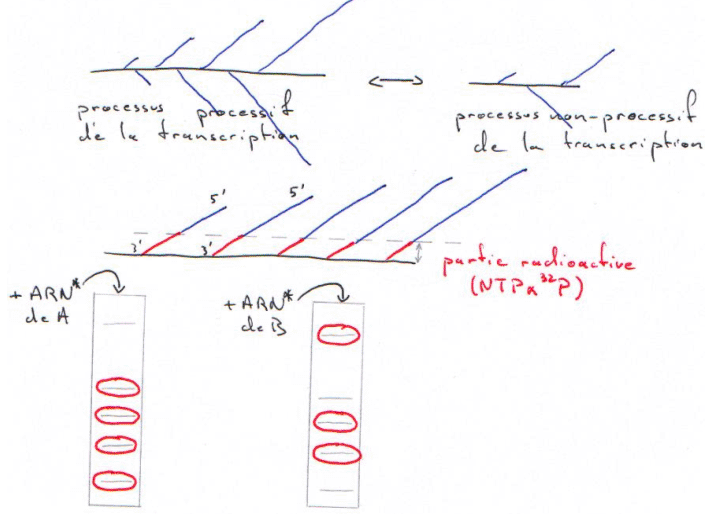
Exemple ; la technique de Run-On.
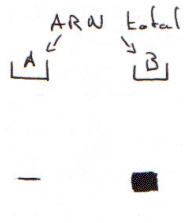
On a hybridé les ARN totaux avec une sonde cDNA X, puis on réalise un northern. On trouve qu’il y a plus d’ARNm en B qu’en A : stabilité des messagers Pour répondre à cette question, on utilise la technique de Run-On.
Cette technique repose sur le passage d’un processus non-processif de la transcription à un processus processif de la transcription. Lorsque l’on isole un noyau pour un gène donné, on fige son état transcriptionnel. In vivo, on lui fait reprendre la transcription (avec des NTP marqués).
On ne ré-initie pas la transcription de l’ARN, on ne fait qu’allonger des chaînes pré-initiées.
D La RNA polymérase III.
Dans une cellule, cette polymérase » transcrit des petits ARN : tRNA, ARN 5s et quelques uRNA.
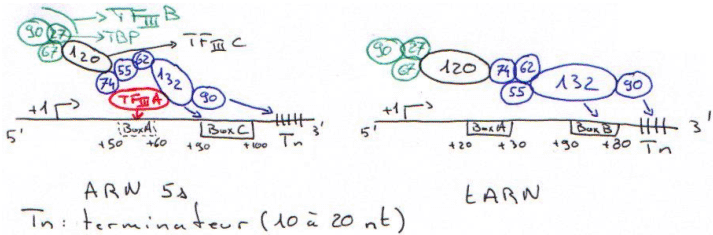
Les promoteurs de la polymérase III sont dans la partie transcrite du gène.
Dans le cas de l’ARN 5s, TFIIIA reconnaît une boite semblable à « boxA ».
TFIIIC recrute TFIIIB puis TBP. Ce dernier n’agit pas avec TCD.
Les RNA polymérases vont se servir d’une matrice ADN et utilisent 4 NTP (nucléotide tri-phosphate). Elles ont besoin d’un site de reconnaissance.
E Les DNAses ou RNAses : les nucléases.
1 Bal 31.
Cette enzyme attaque des deux côtés des brins Elle possède les deux activités : 5’3’nucléase et 3’5’nucléase.
2 Nucléase S1.
Cette enzyme va dégrader l’ADN simple brin (et l’ARN).
3 Nucléase micrococcus.
Cette enzyme digère aussi bien l’ADN que l’ARN.
F La DNA ligase.
Cette enzyme est utilisée pour placer un insert dans un plasmide.
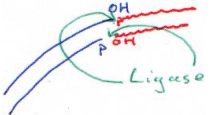
D’abord, on va digérer le plasmide et l’insert par la même ER (par exemple, par BamHI). Pour réaliser cette ligation, il nous faut les deux fragments (plasmide et insert) et la DNA ligase.
En général, on essaie de faire des coupures à extrémités cohésives. En fait, si l’on réalise une coupure qui donne des extrémités franches, la ligase doit rencontrer à la fois le plasmide et l’insert : il y a peu de probabilité de réussite.
Pour obtenir de bons résultats, il faut :
- Se placer en excès de plasmides (100 fois plus).
- Eliminer la possibilité de re-fermeture du plasmide : on applique donc un traitement à la phosphatase. ? La fermeture du plasmide sur lui-même est impossible mais la ligation est toujours réalisable.
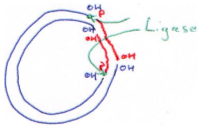
G La reverse transcriptase.
Dans un cas normal, on passe de l’ADN à l’ARN puis aux protéines. Ici, la matrice est l’ARN. La reverse transcriptase va incorporer des dNTP.
Deux activités sont associées à cette enzyme :
- RNAse H : coupe l’hybride ADN/ARN.
- Terminal transférase : ajout de 3 à 5 Cytosine en 3’ si l’ARN en 5’ a subit le capping.
II Les problèmes de marquage de l’ADN.
On peut marquer l’ADN avec une sonde radioactive (32P) ou avec une sonde fluorescente. Ce marquage est effectué soit aux extrémités de la molécule ou bien sur la totalité de celle-ci.
On va utiliser des nucléotides au 32P pour marquer sur tout le long de la molécule ou bien des nucléotides au 32P pour marquer uniquement les extrémités.
Le marquage fluorescent se fait avec des bases fluorescentes.
1 Marquage terminal : le tailing.

2 Marquage terminal : nucléotide kinase et protéine kinase.
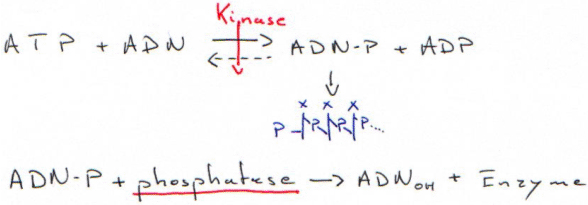
III Le séquençage et la PCR.
Il existe deux méthodes de séquençage : la méthode de Maxam et Gilbert et la méthode de Sanger.
A La méthode de Maxam et Gilbert.
On prépare un tube par type de coupure (une après G, après A, après T, après C). En fonction des concentration d’enzyme et d’ADN, on obtient (statistiquement) une coupure par molécule avec tous les cas différents présents.
B La méthode de Sanger.
Dans ce cas, on va réaliser la synthèse d’ADN avec des arrêts de synthèse, en utilisant des didésoxynucléotides.
L’amorce pour démarrer la synthèse est connue (et vient du commerce). On réalise ensuite la synthèse avec un ddNTP présent.
Cette méthode est valable pour séquencer des brins dont la taille n’excède pas 600 nucléotides.
C La PCR.
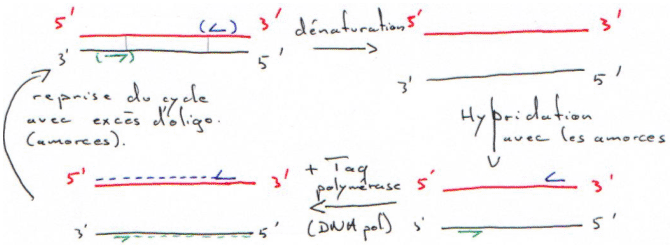
IV Construction d’une banque de cDNA.
En biologie moléculaire, les deux grandes problématiques sont : la structure des gènes et génomes, et, comment sont exprimés les différents gènes dans les divers types cellulaires.
On va dégrader l’ADN par des ER afin d’obtenir des fragments homogènes en taille. On fait entrer ces fragments dans vecteurs, puis, dans des bactéries. On a alors une banque génomique : grand nombre de bactéries ayant intégré un fragment d’ADN (à cloner).
A Les vecteurs.
On a le choix entre différents vecteurs : phages et plasmides. En général, on va utiliser un phage spécial, le phage ? (50 000pb).
1 Le phage .
Le « pili » de la queue du phage reconnaît la bactérie grâce aux récepteurs membranaires et va permettre l’injection de l’ADN dans cette bactérie.
Cet ADN pourra servir dans deux cycles :
- Cycle lysogénique : l’ADN phagique s’insère dans le génome bactérien.
- Cycle lytique : il y a production massive de phages jusqu’à la lyse des bactéries.
L’ADN phagique est transcrit quand il rentre dans la cellule puis se dirige vers une voie ou vers une autre. Dans le cas d’un cycle lytique, il y a synthèse des enzymes nécessaires à la synthèse d’ADN, de la tête et de la queue. La transcription donnera ensuite les protéines de la tête et la fixation sur les sites cos, ce qui permet l’entrée de l’ADN phagique (par aspiration). Il y a arrêt au second site cos. Quand la tête est pleine, la queue vient se fixer.
Sur le phage , la partie permettant la lysogénie fait 15kb. Ce phage est composé d’un bras gauche de 10kb, du droit de 20kb et de la partie centrale « inutile » de 20kb.
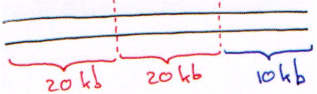
Le choix des ER : les ER utilisées doivent donner des résultats homogènes, avec une taille autour de 15kb. On peut prendre une ER à 4 et on lui fait faire des digestions partielles à 1/50ème.
En réalité, on prend 2 ER et avec chacune d’elle, on effectue les digestions à 1/100ème. Dans ce cas, on aura une distribution homogène.
2 Les plasmides.
Les plasmides ont « Ori » et sont capables de se maintenir dans les bactéries. Leur taille est variable (de 4000pb à 10.000pb).
En 1972, on a vu que les bactéries devaient être compétentes pour réaliser la « transformation bactérienne » : entrée d’ADN extérieur. Les facteurs de compétence sont le phosphate de calcium et la température.
Les plasmides ne seront pas utilisés pour les génomes de grande taille : le rendement de transformation diminue avec la taille (on a le maximum avec 10.000pb).
On va réaliser une combinaison entre le phage et un plasmide.
3 Les cosmides.
Les cosmides sont des vecteurs hybrides portant à la fois :
- des séquences d’origine phagique permettant leur encapsidation in vitro (séquence cos).
- des séquences d’origine plasmidique : gène de résistance à un antibiotique et séquence permettant au plasmide de se répliquer dans une bactérie comme un simple s).
- des séquences d’origine plasmidique : gène de résistance à un antibiotique et séquence permettant au plasmide de se répliquer dans une bactérie comme un simple plasmide (séquence Ori).
Ceci permet de se dispenser des régions essentielles de l’ADN du phage et de construire des ADN recombinants portant jusqu’à 45 kb d’ADN étranger.
Une fois que l’on a placé l’insert (fragment de 45kb), on réalise l’encapsidation et la mise en place de la queue.
Pour avoir un banque complète de cDNA , on prend 10 fois plus de bactéries que de nécessaire.
4 Les YAC (yeast artificial chromosome) ou chromosome de levure.
Ces vecteurs de clonages peuvent se présenter sous deux formes : une forme circulaire, permettant sa manipulation chez E. Coli (et en particulier l’insertion d’ADN exogène et son amplification ils possèdent une séquence ori et un gène de résistance à un antibiotique) et une forme linéaire, obtenue après coupure par BamHI et EcoRI et qui permet d’obtenir les deux “bras” du YAC. Ces deux bras contiennent les télomères (TEL), une origine de réplication levure (ARS) un centromère (CEN) ainsi que des gènes (URA et TRP) utilisés pour la sélection des clones. La ligation de ces bras à un très grand fragment d’ADN humain (100 à 1000 kb) extrait de cellules en culture le transforme en pseudo chromosome de levure capable après introduction dans ce microorganisme de se répliquer (grâce à ARS), de stabiliser ses extrémités (grâce aux séquences TEL) et de se répartir entre les cellules filles lors de la division cellulaire (grâce à la séquence CEN).
Toutefois, ces YAC ont une affreuse tendance à se recombiner avec les chromosomes de la levure. En général, on ajoute entre 2.10^5 et 2.10^6pb.
B Banque de cDNA.
Dans un individu, on isole un type cellulaire et l’on focalise dessus. Pour l’homme, on a entre 20 et 30 cDNA différents.
A partir d’ARNm polyA, on passe au cDNA, que l’on insère ensuite dans un vecteur lequel est transféré dans des bactéries.
1 La reverse transcriptase.
On va hybrider des oligodT (25) sur la queue polyA, c’est ensuite que l’on ajoute la reverse transcriptase et les dNTP. Une fois le travail de la reverse transcriptase achevé, on élimine l’ARN avec la nucléase S1.
On transcrit enfin le brin d’ADN que l’on obtient, mais l’on n’a pas de cDNA pleine taille.
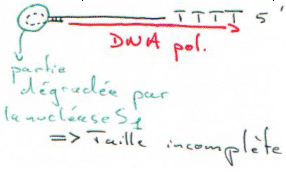
On peut toutefois obtenir des cDNA pleine-taille. Pour cela, on va toujours utiliser la reverse transcriptase. Celle-ci possède les activités :
- RNAse H : elle crée des « nick » dans l’ARN mais avec l’extrémité 3’ portant un OH libre. L’hybride (ARN/ADN) obtenu aura donc un 3’ OH libre. Dans ce cas, on perd aussi un peu d’information en 5’. Le point fort est que l’on contrôle bien la réaction.
- Terminal transférase : On rajoute sur l’hybride un oligonucléotide avec trois G et 20 nucléotides. LA RNAse H va dégrader l’ARN mais pas l’amorce (l’oligo), ce qui permet de faire des cDNA pleine taille.

2 La mise en vecteur.
Les gènes donnent des messagers d’environ 3000nt (de 500 à 10^5nt). Dans ce cas, la taille est correcte pour une insertion dans un plasmide. On trouve deux types de plasmides :
- Les « tout-bête » : ils possèdent une « Ori » et un MCS (pour les ER).
- Les plasmides « vecteurs d’expression » : ils ont une "‘Ori", un MCS et un promoteur. Ils permettent la transcription des inserts, passage en mRNA et puis en protéine mais cette dernière étape est beaucoup plus dure. Pour qu’elle soit possible, il faut que le ribosome trouve un AUG et une séquence consensus (shine-dalgarno) en amont. On doit donc placer sur l’insert une séquence consensus et un AUG.
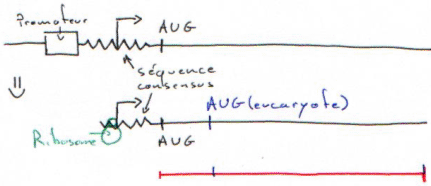
Si le ribosome lit 3/3, il arrive sur AUG et il est en phase : ça marche et l’on a une « protéine de fusion ».
Le nombre de nucléotides avant AUG est différent d’un multiple de 3 et la traduction donnera une fausse protéine.
Le ribosome peut tomber sur un codon stop dans la région non traduite.
La probabilité d’obtenir une protéine sur une banque est faible alors que l’on a du pleine-taille.
Dans le cas d’une taille plus courte, on n’a plus la partie 5’ non codante et l’on a 1 chance sur 3 d’être en phase.
Pour le criblage d’expression, il vaut mieux utiliser des « tailles non entières ».
Dans les fibroblastes, on a environ 30.000 ARNm. On devrait prendre 30.000 bactéries mais on en prend plus car on n’a pas forcément une égalité de répartition des différents ARNm.
On va prendre 100 fois plus de bactéries, sinon, on peut faire des banques normalisées (avec la même quantité de chaque messager).
C Le criblage.
C’est dans l’étape de criblage où l’on fait du clonage.
On étale la banque pour que chaque bactérie soit individualisée : elles donneront des cellules filles identiques (= clonage).
Pour repérer ce qui nous intéresse, il nous faut une sonde nucléotidique et une sonde Anticorps. On pourra identifier la protéine intéressante par spectrométrie de masse (de Malditoff ou de Cuthoff).
Pour la sonde anticorps, on fait faire à un animal, des anticorps contre une protéine (plus l’animal est gros, plus on récupère d’anticorps). Dans 50% des cas, on a des sondes hétérologues (sondes nucléotidiques mais trouvées dans une autre espèce).
La boite contenant les bactéries est répliquée sur un filtre de nitrate de cellulose. On lyse les bactéries répliquées et les macromolécules vont s’hybrider sur les filtres. On ajoute la sonde et on reconnaît (ou non) la présence de cDNA (s’il est présent). On récupère alors, sur la boite, la bactérie qui nous intéresse.
Avec un anticorps, on fait aussi une réplique sur nitrocellulose. La première réaction se fait avec des anticorps de lapin contre les protéines humaines (cDNA humaine). La seconde réaction consiste à rajouter un anticorps anti-lapin fluorescent contre l’anticorps de lapin.
C’est une réaction élisa sandwich.
Chapitre 3 :
La régulation de la transcription.
La transcription est initiée en « +1 » car en amont, le promoteur est reconnu est c’est là où viennent s’installer des complexes d’initiation.
Pour réaliser cette transcription, une cellule va passer par diverses étapes.
La cellule reçoit des informations qui vont entraîner une régulation du génome et donner un système différencié.
Les informations du milieu extérieur vont être intégrées et donner des réponses adaptatives.
On trouve en nombre variable deux séquences portées par l’ADN, situées de part et d’autre du « point +1 » (surtout en amont ce point) : ce sont des séquences de régulation. On a des boites intriquées qui permettent la compétition des divers facteurs de régulation (+ ou -).
I Gènes dont la régulation est cycle cellulaire dépendante.
A Les gènes hormono-régulés.
Les cellules en arrêt de croissance (en phase G0), lorsqu’elles sont stimulées vont mettre en place un programme d’expression génique. Les stimulations peuvent être :
- du sérum,
- des facteurs de croissance (autocrine ou paracrine) : ces derniers sont indispensables mais ils peuvent être cancérigènes.
Le programme d’expression génique va entraîner une transcription précoce ou tardive.
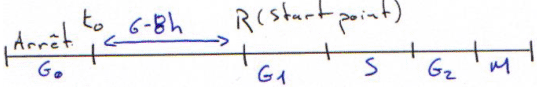
Ces facteurs de croissance permettent de synchroniser une population cellulaire.
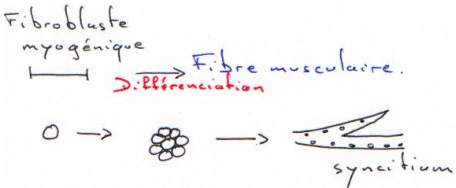
Les hormones stéroïdes reconnaissent des récepteurs intracellulaires. (cf. cours d’endocrinologie).
B Stratégie d’étude ; étude par transfection.
1 Cas généraux.
Dans une cellule eucaryote, on fait entrer des plasmides ou des rétrovirus (infection).
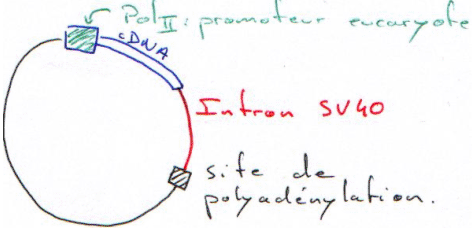
Pour être transcrit, le plasmide doit pouvoir utiliser la machinerie de la cellule hôte.
Les promoteurs (eucaryotes) utilisés peuvent être :
- Fort : haute fréquence d’initiation. Ce sont des promoteurs viraux, de RSV-, CMV (cytomégalovirus), SV40 (virus à ADN qui infecte le singe).
- Faible : faible fréquence d’initiation. Tk, Proliférine.
Le promoteur est choisi en fonction de l’étude que l’on veut faire.
On peut faire fabriquer à des cellules eucaryotes, des produits bactériens (protéines d’intérêt).
Quand les gènes eucaryotes sont transcrits, ils donnent un pré-messager (un chacun) (quand il est transcrit par la polymérase II). Ce dernier est reconnu comme pré-messager et va subir une maturation. Ce messager est ensuite dirigé vers le cytoplasme pour permettre la synthèse de protéine.
Il y a une relation entre la quantité de protéines produites et la quantité de transcription.
2 Transfections réalisées.
2.1 Transfection transitoire.
La cellule après la transcription, est utilisée dans un temps très court pour éviter l’intégration.
On utilise ce système pour étudier la régulation des promoteurs.
Le cDNA code pour un gène rapporteur (protéine facilement repérable, de procaryotes). On trouve :
- CAT : Chloramphénicol Acétyl Transférase (La CAT transforme le chloramphénicol en forme inactive).
- -galactosidase.
- Luciférase.
On réalise un broyat cellulaire auquel on ajoute de l’acétate (sous forme d’ACo A) et du chloramphénicol. Si ce dernier est acétylé, c’est que l’enzyme est présente.
Une cellule eucaryote peut intégrer 1 à 50 plasmides. Attention, deux populations cellulaires peuvent intégrer un nombre différent de plasmides (le volume de synthèse sera différent). On ajoute donc un plasmide de contrôle ou un gène constitutif non régulé (comme la -gal) dans le plasmide.
Pour obtenir le standard interne, on fait le rapport : Activité CAT/Activité -gal.
Au bout d’un moment (72 heures), tous les plasmides non intégrés vont être dégradés. Il faut attendre entre 24 et 48 heures pour que le plasmide s’installe.
On a donc entre 1 et 2 jours pour travailler.
2.2 Transfections stables.
Il y a introduction définitive d’un gène (plasmide) dans le génome.
La fréquence de réussite est de 10-5 à 10-6.
Pour crible positif, on ajoute sur le plasmide, un gène de résistance à la néomycine (G418) et on réalise les cultures en présence de l’antibiotique spécifique.
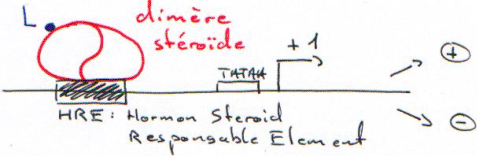
3 Les séquences reconnues par les récepteurs.
Ces séquences sont des séquences bipartites.

Ces séquences présentent des homologies (en rouge).
Sous forme de dimère, le récepteur reconnaît la séquence et selon celle-ci, il répresse ou active.

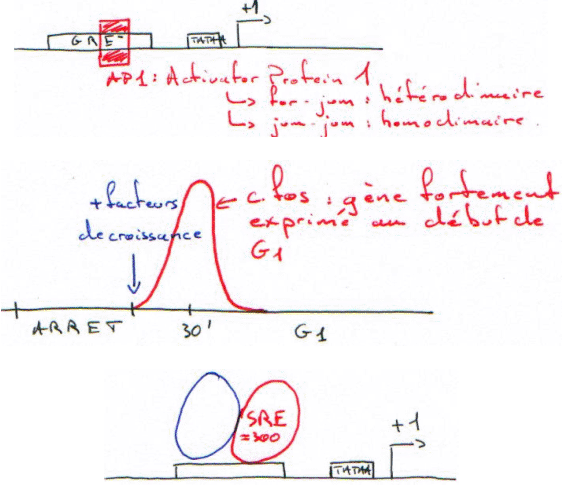
Les gènes responsables de la transformation des cellules sont des oncogènes : si le gène est (trop) stimulé, on arrive au stade de cellule cancéreuse.
A chaque oncogène viral (V-Fos) correspond un gène chez les mammifères : les proto-oncogènes (C-Fos). Ces derniers sont les gènes assurant la régulation du cycle et les effecteurs (réplication et transcription).
Exemple de la protéine Sarc :
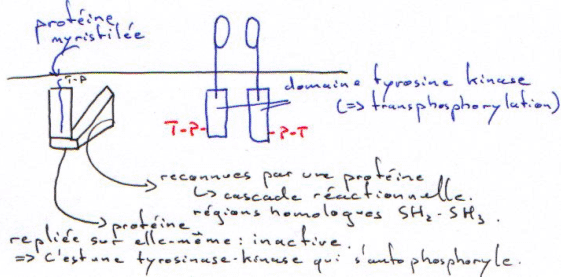
Quand le Tyr-P (T-P sur le schéma) du dimère vient sur la protéine myristilée, il y a ouverture de l’enzyme et activation de celle-ci. Une mutation ponctuelle peut entraîner la forme virale, toujours ouverte.
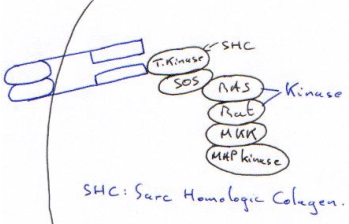
La MAP-Kinase va phosphoryler ce qui est sur SRE (avant la tata box). Dès que le ligand se fixe, il y a endocytose du complexe c’est une période réfractaire.
C’est l’activation transitoire de la kinase.
D’une façon générale, la kinase et la phosphatase sont associées au site de phosphorylation. A t=x, si la phosphatase est plus active que la kinase, on obtient une protéine de phosphorylation (l’inverse est vrai).
Dans le noyau, une phosphatase est constitutive par un jeu de phosphorylation par la déphosphatase On peut expliquer l’activation de C-Fos.
Remarque : L’activation de C-Fos est quasi immédiate. Ce système évite de synthétiser de nouvelles molécules. Il n’y a qu’à phosphoryler les protéines constitutives pour les activer. C’est l’étape précoce.
Il y a ensuite synthèse d’AP1 qui va stimuler tous les gènes qui le reconnaissent. ? C’est l’étape tardive de synthèse.
Il y a un point R (de restriction) entre les deux phases (précoce/tardive). Le passage de ce point permet la prolifération.

